Hochleistungsflüssigkeitschromatographie
Titelblatt
Bericht der Expertengruppe für
Hochleistungs-Flüssigkeitschromatographie
SoSe 2021
Abgabedatum
21.06.2021
Über-/Expertengruppe 05
Anna Bauer
Christine Ewig
Franziska Heich
Jolien Kürschner
Yana Moser
Undine Welle
Hochleistungs-Flüssigkeitschromatographie
Inhaltsverzeichnis
Einleitung
Die Hochleistungs-Flüssigkeitschromatographie (high performance liquid chromatography, kurz: HPLC) wird hauptsächlich, im Zuge von Identitäts- und Reinheitsprüfung und Gehaltsbestimmung, als Methode zur analytischen Stofftrennung angewendet. Zudem kann sie zu präparativen Zwecken genutzt werden. Sie zählt zu den äußeren chromatographischen Trennmethoden, bei der alle Substanzen die gleiche Wegstrecke zwischen Injektor und Detektor zurücklegen und die dafür benötigte Zeit (Retentionszeit) gemessen wird.
Grundlagen (physikalische/chemische)
Bei der HPLC handelt es sich um eine Variante der Säulenchromatographie. Sie beruht auf Wechselwirkungen zwischen dem Analyten, welcher mit der mobilen Phase transportiert wird, und der stationären Phase. Bei der stationären Phase handelt es sich um eine feste Oberfläche in der Trennsäule, die abhängig von den zu untersuchenden Substanzen in verschiedenen Variationen vorzufinden ist. Die Substanzen der mobilen Phase besitzen unterschiedliche Affinitäten zu der stationären Phase und lassen sich somit gut trennen. Dabei zeichnen sich Stoffe mit hoher Affinität durch eine verlängerte und Stoffe mit einer niedrigeren Affinität durch eine verkürzte Retentionszeit innerhalb der Trennsäule aus.
Mithilfe von Pumpen wird das Elutionsmittel durch die Trennsäule gedrückt. Da die stationäre Phase aus sehr kleinen Partikeln besteht, ist die Packungsdichte in der Trennsäule im Vergleich zu anderen flüssigchromatographischen Verfahren, bei denen größere Partikel verwendet werden, sehr hoch. Aus der hohen Packungsdichte resultiert ein hoher Fließwiderstand. Folglich kann eine ausreichende Strömungsgeschwindigkeit nur mithilfe von Hochdruckpumpen erreicht werden, die Schwerkraft ist nicht ausreichend. Der notwendige Druck hängt neben der Packungsdichte auch von der Länge der Trennsäulen und der Viskosität des Elutionsmittels ab. Die Trenneffizienz wird durch die Partikelgröße und der Partikelgrößenverteilung beeinflusst.
Heutzutage ist die C-18-Umkehrphase, bei der Octadecylgruppen in die stationäre Phase eingeführt werden, eine der relevantesten Methoden im pharmazeutischen Bereich. Mithilfe der HPLC sind fast alle löslichen organischen und anorganischen Arzneistoffe trennbar, da eine Vielzahl an Kombinationsmöglichkeiten von mobiler und stationärer Phase möglich sind. Es kann nahezu jede gewünschte Selektivität eingestellt werden.1 2
Trennmethoden
Die Trennung bei der Normalphasenchromatographie basiert auf unterschiedlichen Adsorptionsverhalten, sprich Anhaftungs- und Ablöseverhalten der Probe an der stationären Phase. Dies bedeutet, dass Probenmoleküle durch Wechselwirkungen mit der polaren stationären Phase zeitweilig an dessen Oberfläche angeheftet sind, durch Verteilungsvorgänge aber auch wieder abgelöst und mit dem Eluenten zum Detektor transportiert werden können. Je stärker die polaren Eigenschaften der Probenbestandteile sind, desto besser interagieren sie mit der polaren stationären Phase. Demzufolge verbleiben Substanzen mit hoher Polarität länger in der Trennsäule. Als Elutionsmittel sind möglichst unpolare Substanzen einzusetzen, da eine starke Wechselwirkung mit der stationären Phase zu einer Verdrängung der Probe und somit Verkürzung der Retentionszeit führt. Dies hat eine verminderte Trennleistung als Folge. Die Elutionskraft beschreibt die Fähigkeit eines Eluenten, Probenmoleküle von der stationären Phase in die mobile Phase zu überführen, bezogen auf das Kieselgel. Polare Fließmittel haben demzufolge eine große Elutionskraft und bewirken eine kurze Retentionszeit der Analyten.
Nach diesen Betrachtungen wurde die elutrope Reihe aufgestellt,
welche die empirische Anordnung der Fließmittel nach steigender Elutionskraft
darstellt. Begonnen wird mit dem unpolarsten Fließmittel, also mit dem
Fließmittel der niedrigsten Elutionskraft:
Hexan < Cyclohexan < Benzen < Chloroform < Dichlormethan < Diethylether <
Ethylacetat < Aceton < Dioxan < Acetonitril < Methanol < Wasser 3 4
5 6
Die Umkehrphasenchromatographie (RP) beruht auf Interaktionen zwischen den in
die stationäre Phase eingeführten Alkylresten und den Substanzproben, in Form
von Adsorptions- und Verteilungsvorgängen. Die Umkehrphase spielt eine große
Rolle in der pharmazeutischen Analytik. Im Gegensatz zu der
Normalphasenchromatographie wird sie vor allem bei der Trennung von unpolaren
Substanzen angewendet. Als mobile Phase werden polare Fließmittel, häufig
Acetonitril oder Methanol-Wasser-Gemische, eingesetzt. Die Elutionsreihe,
demzufolge auch die elutrope Reihe der Fließmittel ist hier in Bezug zur Normalphase umgekehrt.
Je nach Derivatisierung der freien Hydroxylgruppen an den Silanolgruppen des
Kieselgels lassen sich die Bindungseigenschaften der stationären Phase anpassen.
Die Alkylierung wird mithilfe von Organochlorsilanen durchgeführt.
Die Modifikation läuft dabei in zwei Schritten ab:
Zunächst durch die Silanisierung, bei
der raumfordernde Alkylgruppen, zum Beispiel Octyl- (RP-8) und Octadecylgruppen
(RP-18, ODS), eingeführt werden.
Folgend aus dem Endcapping, bei dem
Trimethylchlorsilane zur Derivatisierung der verbleibenden Silanolgruppen
eingeführt werden. Diese Modifikation führt zu einem stark lipophilen Charakter,
wodurch vermehrt Interaktionen mit unpolaren Molekülen auftreten. Durch
Einführen von Abstandshalter (Spacer) können die Abstände zwischen den
funktionellen Gruppen beeinflusst werden. Dabei handelt es sich häufig um
lipophile Gruppen, wie z.B. Alkyl-, Cyclohexyl-, oder Phenylreste. Dies wird
gemacht, um spezifische Wechselwirkungen mit den zu trennenden Substanzen zu
erzielen.7 8 9
Die Größenausschlusschromatographie (SEC) folgt dem Trennverfahren nach unterschiedlichen Molekülgrößen der Analyten und wird oft bei Proteinen und Polymeren angewendet. Die Oberfläche der stationären Phase besteht aus porösen Polymeren oder Kieselgel. Kleine Moleküle können tiefer in die Poren eindringen und eluieren somit langsamer als große Moleküle. Die mobile Phase dient in dieser Trennmethode ausschließlich als Transportmittel und besteht meist aus organischen Lösemitteln oder Pufferlösungen. Dementsprechend sollte sie keine Wechselwirkung mit der stationären Phase aufweisen und gut löslich in dieser sein. Der lineare Bereich der Eichkurve, ein Mittel zur graphischen Darstellung des Zusammenhangs zwischen der Elutionszeit und der Molekülgröße, gibt Aufschluss über die geeignete Wahl des Packungsmaterials mit entsprechender Porengröße der stationären Phase.10 11 12
Die Ionenchromatographie (IC) findet vor allem bei kleineren organischen und anorganischen Ionen Anwendung. Die IC folgt dem Ionenaustausch-Prinzip, das heißt, dass die Pufferionen der mobilen Phase mit den Ionen des Analyten um die Austauschplätze an der stationären Phase konkurrieren. Dieser Wettstreit tritt auf, da ionische Wechselwirkungen der ionisierbaren funktionellen Gruppen an der stationären Phase (beschichtete Polymere oder Kieselgel) ausgenutzt werden. Die stationäre Phase wird durch die pH-Eigenschaft des Elutionsmittels ionisiert, wobei sich für eine Anionentrennung oft ein Natriumhydrogencarbonat-Puffer eignet und für eine Kationentrennung ein Wasserstoffchlorid-Puffer. Bei dieser Trennmethode kann die Elutionsreihenfolge für An- und Kationen berücksichtigt werden.13 14 15
Die Affinitätschromatographie dient der Isolierung von Substanzen aus biochemischen Gemische mit großer Spezifität, indem biospezifische Wechselwirkungen, wie z.B. Antigen/Antikörper, Enzym/Inhibitor oder auch komplementärer Nukleinsäurepaare, ausgenutzt werden. Hierbei bindet eine entsprechende Substanz aus der Probe selektiv an spezifischen Molekülen, sogenannte Liganden, die an einen unlöslichen Träger (Polymere oder Kieselgel) angebracht sind. Dieser wird durch Komplexierung an die stationäre Phase gebunden, während andere Begleitsubstanzen der Probe von der mobilen Phase getrennt bzw. heruntergespült werden. Die Elution des Analyten erfolgt durch ein Pufferwechsel, die mobile Phase wird z.B. durch einen Salzgradienten, pH-Gradienten oder einer Probenkonkurrenz verändert und löst den Analyten vom Liganden, was zu einem aufgereinigten Protein führt.16 17 18
Parameter
Bei einer HPLC spielen einige chromatografische Parameter eine Rolle. Diese werden im Nachfolgenden erläutert.
Retentionszeiten

⚠ $t_M=t_0$: Totzeit
Beschreibt die Zeit von der Injektion der Probe bis zum ersten Signal, welches durch das Lösemittel ausgelöst wird. Hier findet noch keine Interaktion des Analyten mit der stationären Phase statt, es ist die reine Passierzeit der mobilen Phase.
⚠ $t_R$: Bruttoretentionszeit
Zeit von der Injektion der Probe bis zum Probensignal.
⚠ $t_S$: Nettoretentionszeit
Zeit vom Lösemittelsignal bis zum Probensignal.
⚠ $t_S= t_R- t_M$
⚠ $AUC$: „Area under curve“
Beschreibt die durch Integralrechnung berechnete Fläche unter dem Peak. Diese gibt Aussage über die Quantität des/der Analyten.
Durchflussrate: ⚠ $F$ in ml/min
Totvolumen: ⚠ $V_M= t_M · F$
Volumen, welches das System von Injektion bis zum Detektor durchfließt, ohne mit der Säule zu interagieren.
Retentionsvolumen: ⚠ $V_R= t_R · F$
Volumen, welches das System von Injektion bis zum Detektor durchfließt, nachdem mit der stationären Phase interagiert wurde.
Retentionsfaktor: ⚠ $k= \frac{t_R-t_M}{t_M} = \frac{t_S}{t_M}$
Der Retentionsfaktor ⚠ $k$ gibt Auskunft über das Verhältnis zwischen Aufenthalt der Substanz in stationärer und mobiler Phase. Er ist ein Maß für die Wanderungsgeschwindigkeit in einem chromatographischen System, unabhängig von der Säulenlänge und der Fließgeschwindigkeit, und somit ein genauerer Parameter als die Nettoretentionszeit alleine.20
Verteilungskoeffizient ⚠ $K$
⚠ $K= \frac{Konzentration_{stationäre Phase}}{Konzentration_{mobile Phase}}$
Der Verteilungskoeffizient ⚠ $K$ beschreibt einen theoretischen Gleichgewichtszustand zwischen der Konzentration des Analyten und der Konzentration der stationären Phase bei gleichen Volumina. Bei verschiedenen Verteilungskoeffizienten von einzelnen Analyten kommt es zur Trennung.21
Trennfaktor/Selektivitätsfaktor ⚠ $α$
⚠ $α= \frac{k_2}{k_1}$
⚠ $k_2$ : später eluierter Peak
⚠ $k_1$ : früher eluierter Peak
Der Trennfaktor ⚠ $α$ gibt das Verhältnis zwischen zwei Retentionszeiten an.
Je größer die Differenz der Retentionszeiten ist, umso einfacher gelingt eine Trennung.
Die Werte liegen in der Regel zwischen 1 und 10, wobei Werte im höheren Bereich zu bevorzugen sind.22
Symmetriefaktor ⚠ $A_S$
⚠ $A_S= \frac{w_{0,05}}{2d}$
⚠ $w_{0,05}$: Breite des Peaks in einem Zwanzigstel der Peakhöhe
⚠ $d$: Peakbreite des aufsteigenden Astes auf Höhe von einem Zwanzigstel bis zur Mitte des Peaks.
Der Symmetriefaktor ⚠ $A_S$ gibt Auskunft über die Form eines Peaks.

Wenn:
⚠ $A_S=1$ : Symmetrie
⚠ $A_S>1$ : Tailing
⚠ $A_S<1$ : Fronting
Das Ph.Eur. gibt Werte von 0,8 – 1,5 an.24
Auflösung ⚠ $R_S$
Die Auflösung ⚠ $R_S$ ist ein Maß für die Fähigkeit eines chromatographischen Systems, zwei Analyten so zu trennen, dass eine Auswertung vorgenommen werden kann.
⚠ $R_S= 1,18 · \frac{t_{R2} - t_{R1}}{w_{h1}+w_{h2}}$
Die Auflösung ⚠ $R_S$ sollte mindestens 1,5 betragen, ansonsten gehen die Peaks ineinander über und es findet keine Basislinientrennung statt.
⚠ $t_{R1}$: Bruttoretentionszeit des früher eluierten Analyten
⚠ $t_{R2}$: Bruttoretentionszeit des später eluierten Analyten
⚠ $w_{h1}$: Peakbreite des früher eluierten Analyten auf halber Peakhöhe
⚠ $w_{h2}$: Peakbreite des später eluierten Analyten auf halber Peakhöhe 25

Peak-Tal-Verhältnis ⚠ $p/v$
⚠ $p/v= \frac{H_p}{H_v}$
⚠ $H_p$: Höhe des kleineren Peaks
⚠ $H_v$: Höhe der Überschneidung beider Peaks bis zur Basislinie.
Das Peak-Tal-Verhältnis wird als Alternative zur Auflösung ⚠ $R_S$ herangezogen, wenn zwischen zwei Peaks keine Basislinientrennung erkennbar ist, um das Verhältnis zweier Peaks zueinander darzustellen. Somit kann beurteilt werden, wie gut beide Peaks voneinander unterschieden werden können.27
Signal-Rausch-Verhältnis ⚠ $S/N$
⚠ $S/N= \frac{2H}{h}$
⚠ $H$: Peakhöhe des Analyten
⚠ $h$: Höhe des Rauschens; wird ermittelt über mind. die fünfmalige Breite der Signalpeakbreite (⚠ $w_h$) auf halber Höhe.
Das Signal-Rausch-Verhältnis gibt Auskunft darüber, wie gut man einen Peak vom Grundrauschen der Basislinie unterscheiden kann.
Detektionsgrenze (⚠ $LOD$= limit of detection): ⚠ $S/N ≥ 3$
Bestimmungsgrenze (⚠ $LOQ$= limit of quantification): ⚠ $S/N ≥ 10$
Wiederholpräzision
Durch die Wiederholpräzision erfolgt die Bestimmung der Reproduzierbarkeit einer Messung. Sie ist die relative Standardabweichung in % bei mindestens drei aufeinanderfolgenden Injektionen. Die dadurch erhaltenen Einzelwerte müssen unter den gleichen Versuchsbedingungen gemessen werden (Messverfahren, Temperatur, Ort, Gerät usw.).
⚠ $S_r (\%) = \frac{100}{\bar{x}}\cdot\sqrt{\frac{\sum{(x-\bar{x})^2}}{n-1}}$
⚠ $xi$: Einzelwerte der Messungen in Form von Peakhöhe, Peakfläche oder dem Verhältnis von Peakflächen
⚠ $x̅$: Mittelwert der Einzelwerte
⚠ $n$: Anzahl der Einzelwerte 29
Trennstufenzahl ⚠ $N$
Eine Trennstufe ist die abgeschlossene Verteilung zwischen stationärer und mobiler Phase. Die Trennstufenzahl ⚠ $N$ ist eine Größe, die eine Aussage über die Trennleistung gibt. Sie ist abhängig von der Retentionszeit einer Substanz, also vom Analyten und dem Messgerät. Je mehr Einzeltrennungen erfolgen, also je höher ⚠ $N$, umso besser ist die Trennleistung der Säule.30
⚠ $N= 5,54 · \left(\frac{t_R}{w_h}\right)²$
Trennstufenhöhe ⚠ $H$
Die Trennstufenhöhe ⚠ $H$ ist eine weitere Größe, die eine Aussage über die Trennleistung gibt.
⚠ $H= \frac{L}{N} = A + \frac{B}{u} + C · u$
⚠ $L$: Säulenlänge in cm
Je kleiner ⚠ $H$, umso besser ist die Trennleistung der Säule.31
Van-Deemter-Gleichung
 Abb.4: Van-Deemter-Gleichung 32
Abb.4: Van-Deemter-Gleichung 32Die Van-Deemter-Gleichung gibt Aufschluss darüber, wie die Trennleistung von der Strömungsgeschwindigkeit (⚠ $u$) der mobilen Phase beeinflusst wird.
⚠ $H= \frac{L}{N}= A + \frac{B}{u} + C · u$
⚠ $u= \frac{L}{t_m}$
Mehrwegeffekt (Eddy-Diffusion) ⚠ $A$:
beschreibt den Weg bzw. die Umwege, die der Analyt durch die stationäre Phase zurücklegen muss (unabhängig von ⚠ $u$).
Längs-/Longitudinaldiffusion ⚠ $B$:
der Analyt bewegt sich nicht nur in direkter Strömungsrichtung, sondern diffundiert zum Beispiel auch entgegengesetzt dieser. Wird die Strömungsgeschwindigkeit ⚠ $u$ erhöht, steigt die Tendenz zur Bewegung in Strömungsrichtung (antiproportional zu ⚠ $u$).
Massenübergang ⚠ $C$:
der Massenübergang beschreibt die Interaktion des Analyten mit der stationären Phase, bei der die Moleküle des Analyten in Poren der stationären Phase aufgehalten und somit erst später wieder in die mobile Phase übergehen. Dies führt zu einer Verbreiterung des Peaks (proportional zu ⚠ $u$).33
Instrumenteller Aufbau

Bei der HPLC-Anlage wird die mobile Phase durch ein Pumpensystem aus dem Flüssigkeitsreservoir durch das Probeninjektionssystem, über die Trennsäule zu dem Detektor gepumpt (s. Abb 5). Die Probelösung wird über das Injektionssystem in den Flüssigkeitsstrom eingebracht und gelangt ebenfalls über die Trennsäule zum Detektor. Die Trennsäule ist dabei bepackt mit der stationären Phase. Das Detektorsignal wird meist computergestützt ausgewertet. Bei manchen Analysen ist ein Gradientenmischer notwendig, um die Zusammensetzung des Eluentengemisches während einer Trennung zu verändern.35
Eluent
Die Elutionsmittel sollten bei der HPLC sorgsam ausgewählt werden. Entscheidend sind hierbei vor allem der Trennmechanismus und die Eigenschaften der zu trennenden Substanzen. Beispielsweise verwendet man in der Normalphasenchromatographie häufig ein Elutionsmittel mit geringer Polarität, in der Umkehrphasenchromatographie hingegen eine wässrige, polare mobile Phase (s. Grundlagen).
Die Qualität der verwendeten Eluenten ist sehr wichtig, eine hohe Reinheit wird vorausgesetzt. Dies bedeutet, dass die Elutionsmittel frei von Gasen sowie unlöslichen Verunreinigungen und Schwebstoffen sein sollten. Um dies zu gewährleisten, sollte der Eluent vor der Verwendung entgast und durch eine engporige Fritte filtriert werden.
Der Eluent darf kein Störsignal in dem verwendeten Detektor verursachen.36
Pumpen
Bei der HPLC werden Kolbenpumpen verwendet, die ein oder mehrere Kolben zur Beförderung der Eluenten besitzen. Da die Pumpen hohen Drücken von 300-400 bar standhalten müssen, werden sie aus widerstandsfähigen Materialien hergestellt. Bei den Kolben wird meist Saphir verwendet, die Pumpenköpfe werden aus Edelstahl, Titan oder Keramik angefertigt. Bei vielen in der HPLC einsetzbaren Detektor ist ein pulsationsarmer, kontinuierlicher Fluss des Elutionsmittels nötig, um Signalschwankungen zu vermeiden. Dies kann durch Verwendung mehrerer Pumpen und gegebenenfalls durch Nachschaltung eines Dämpungselements erreicht werden. Zum Schutz der Pumpe sollte ein minimaler und ein maximaler Druck festgelegt werden, um sowohl das Trockenlaufen, als auch zu hohe Druckanstiege bei Verstopfungen zu vermeiden. Außerdem sollten die Pumpen nach Verwendung gründlich gereinigt werden.37
Injektionssystem
Die Probe wird mit einer Spritze in die Dosierschleife gegeben. Mit dem Sechswegeventil wird entweder "Ladestellung" oder "Injektionsstellung" gewählt. In der Ladestellung fließt der Eluent von der Pumpe zur Trennsäule. In der Injektionsstellung wird das Elutionsmittel durch die Dosierschleife umgeleitet, dabei gelangt die Probe zur Trennsäule.38
Trennsäule und stationäre Phase
Trennsäulen werden meist aus rostfreiem Stahl, aber auch aus Glas oder stahlummanteltem Glas hergestellt. Sie können von 5 bis 30 cm lang sein und haben meist einen Durchmesser von 2 bis 8 mm.
Befüllt ist die Trennsäule mit der stationären Phase. Um eine gute Trennleistung zu erreichen, sollten Teilchen mit möglichst kleinen Durchmessern verwendet werden. Allerdings gibt es eine untere Grenze, da der Druckabfall innerhalb der Säule zunimmt, je kleiner die Teilchen sind. Durchschnittlich liegt der Teilchendurchmesser zwischen 2 und 15 µm.
Eine große Oberfläche der Teilchen ist entscheidend für die Interaktion zwischen stationärer und mobiler Phase. Deshalb werden Trennstoffe mit poröser Oberfläche, wie Kieselgel und Aluminiumoxid, bevorzugt eingesetzt. Der Porendurchmesser beträgt dabei durchschnittlich 7-10 nm.
Trennstoffe können je nach Anwendungsgebiet chemisch modifiziert oder beschichtet werden. Dies ist vor allem bei der Umkehrphasenchromatographie relevant, da bei dieser nicht mit der ursprünglichen, hydrophilen Oberfläche der stationären Phase gearbeitet wird, sondern mit einer Hydrophoben. Um dies zu erreichen werden die Silanolgruppen des Kieselgels mithilfe von Alkylchlorsilanen umgesetzt (s. Trennmethoden).
Eine Modifikation wird auch bei der Trennung chiraler Substanzen benötigt, hierfür werden Cyclodextrine oder Amylosederivate an die Silanolgruppen des Kieselgels gebunden.39
Detektoren
Bei der HPLC können, je nach Substanz- und Eluenteneigenschaften, eine Vielzahl von Detektoren verwendet werden.
UV/Vis- und Fluoreszenzdetektoren eignen sich besonders gut aufgrund ihrer hohen Empfindlichkeit. Voraussetzung für diese Methode ist, dass der Eluent keine Eigenabsorption im betreffenden Bereich aufweist. Zudem muss der Analyt Strahlung im UV/Vis-Bereich absorbieren und bei der Fluorimetrie dies unter Fluoreszenz abgeben. Dies kann auch durch eine Derivatisierung erzielt werden. Erfolgt diese als Nachsäulenderivatisierung, so spricht man von einem chemischen Reaktionsdetektor.
Bei der Anwendung von Brechungsindexdetektoren ist zu beachten, dass diese sehr empfindlich auf Veränderungen von Druck, Temperatur und Lösungsmittelverhältnis reagieren.
Leitfähigkeitsdetektoren werden für Substanzen eingesetzt, die in Ionenform vorliegen, hingegen finden elektrochemische Detektoren, vor allem voltametrische oder amperometrische Verfahren, bei oxidierbaren oder reduzierbaren Substanzen Verwendung.
Der Einsatz von massenselektiven Detektoren bringt einige Schwierigkeiten mit sich, da sich zum einen nicht alle Substanzen leicht in die Gasphase überführen lassen und zum anderen die große Flüssigkeitsmenge in der HPLC nach dem Verdampfen zum Zusammenbrechen des Hochvakuums führt.40
Gradientenmischer
Wenn eine optimale Trennung der Analyten die Veränderung der Mischungsverhältnisse verschiedener Eluenten voraussetzt, wird ein Gradientenmischer genutzt. Man unterscheidet allgemein zwischen Stufengradienten und stufenlosen Lösungsmittelgradienten.
Bei Stufengradienten, die relativ einfach zu erzeugen sind, wird in bestimmten Zeitabständen das Eluentmengenverhältnis geändert. Stufenlose Gradienten werden wiederum in Niederdruck- und Hochdruckgradienten unterteilt. Der Niederdruckgradient wird vor der HPLC-Pumpe mit Hilfe eines Systems von Schaltventilen gebildet. Verschiedene Lösungsmittel werden dadurch in eine Mischkammer befördert, deren Öffnungszeiten, je nach benötigtem Mischungsverhältnis, durch ein Programm geregelt werden. Die Erzeugung von Hochdruckgradienten ist aufwendiger, liefert aber auch sehr genaue Ergebnisse. Der Gradient wird im Hochdrucksystem erzeugt, deshalb wird für jede Lösungsmittelkomponente eine eigene HPLC-Pumpe benötigt.41
Einzelnachweise
1 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013), S.472ff. ⇑
2 Dominik, Steinhilber, Wurglics: Instrumentelle Analytik kompakt, Wissenschaftliche Verlagsgesellschaft Stuttgart, 3. Aufl. (2013) S.243ff. ⇑
3 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/croma/hplc_detail1.vlu/Page/vsc/de/ch/3/anc/croma/hplc/stat_phase/statphase_hplc2.vscml.html (18.05.2021) ⇑
4 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.475f. & S.487. ⇑
5 Skript Instrumentelle Analytik, Nadine Francke ⇑
6 Dominik, Steinhilber, Wurglics: Instrumentelle Analytik kompakt, Wissenschaftliche Verlagsgesellschaft Stuttgart, 3. Aufl. (2013) S.218ff. ⇑
7 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.480 ff & S.488. ⇑
8 Dominik, Steinhilber, Wurglics: Instrumentelle Analytik kompakt, Wissenschaftliche Verlagsgesellschaft Stuttgart, 3. Aufl. (Jahr) S.219 ff. ⇑
9 Skript Instrumentelle Analytik, Nadine Francke ⇑
10 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.491ff. ⇑
11 Skript Instrumentelle Analytik, Nadine Francke ⇑
12 Dominik, Steinhilber, Wurglics: Instrumentelle Analytik kompakt, Wissenschaftliche Verlagsgesellschaft Stuttgart, 3. Aufl. (2013) S.201f. & S.224ff. ⇑
13 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.490f. ⇑
14 Skript Instrumentelle Analytik, Nadine Francke ⇑
15 Dominik, Steinhilber, Wurglics: Instrumentelle Analytik kompakt, Wissenschaftliche Verlagsgesellschaft Stuttgart, 3. Aufl. (2013) S.221ff. ⇑
16 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.493 ⇑
17 Skript Instrumentelle Analytik, Nadine Francke ⇑
18 Dominik, Steinhilber, Wurglics: Instrumentelle Analytik kompakt, Wissenschaftliche Verlagsgesellschaft Stuttgart, 3. Aufl. (2013) S.226f. ⇑
19 Skript Instrumentelle Analytik, Nadine Francke; (Eigene Zeichnung) ⇑
20 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.492 ⇑
21 Eberhard Ehlers: Analytik II, Kurzlehrbuch Quantitative und instrumentelle pharmazeutische Analytik, Deutscher Apotheker Verlag Stuttgart, 11. Aufl. (2008) S.480, 507 ⇑
22 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.430f. ⇑
23 Skript Instrumentelle Analytik, Nadine Francke; (Eigene Zeichnung) ⇑
24 Skript Instrumentelle Analytik, Nadine Francke ⇑
25 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.431ff. ⇑
26 Skript Instrumentelle Analytik, Nadine Francke; (Eigene Zeichnung) ⇑
27 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.433f. ⇑
28 Skript Instrumentelle Analytik, Nadine Francke ⇑
29 Skript Instrumentelle Analytik, Nadine Francke ⇑
30 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.424ff. ⇑
31 Skript Instrumentelle Analytik, Nadine Francke ⇑
32 Skript Instrumentelle Analytik, Nadine Francke; (Eigene Zeichnung) ⇑
33 Skript Instrumentelle Analytik, Nadine Francke ⇑
34 Eberhard Ehlers: Analytik II, Kurzlehrbuch Quantitative und instrumentelle pharmazeutische Analytik, Deutscher Apotheker Verlag Stuttgart, 11. Aufl. (2008) S. 520f.; (Eigene Zeichnung) ⇑
35 Eberhard Ehlers: Analytik II, Kurzlehrbuch Quantitative und instrumentelle pharmazeutische Analytik, Deutscher Apotheker Verlag Stuttgart, 11. Aufl. (2008) S.520ff. ⇑
36 Skript Instrumentelle Analytik, Geräteaufbau, Nadine Francke ⇑
37 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.477 ⇑
38 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.478f. ⇑
39 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.479ff. ⇑
40 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden, Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.483ff. ⇑
41 Rücker, Neugebauer, Willems: Instrumentelle pharmazeutische Analytik, Lehrbuch zu spektroskopischen, chromatographischen, elektrochemischen und thermischen Analysenmethoden; Wissenschaftliche Verlagsgesellschaft Stuttgart, 5. Aufl. (2013) S.477 ⇑
Monographiebeispiel: Tinidazol, Prüfung auf Reinheit
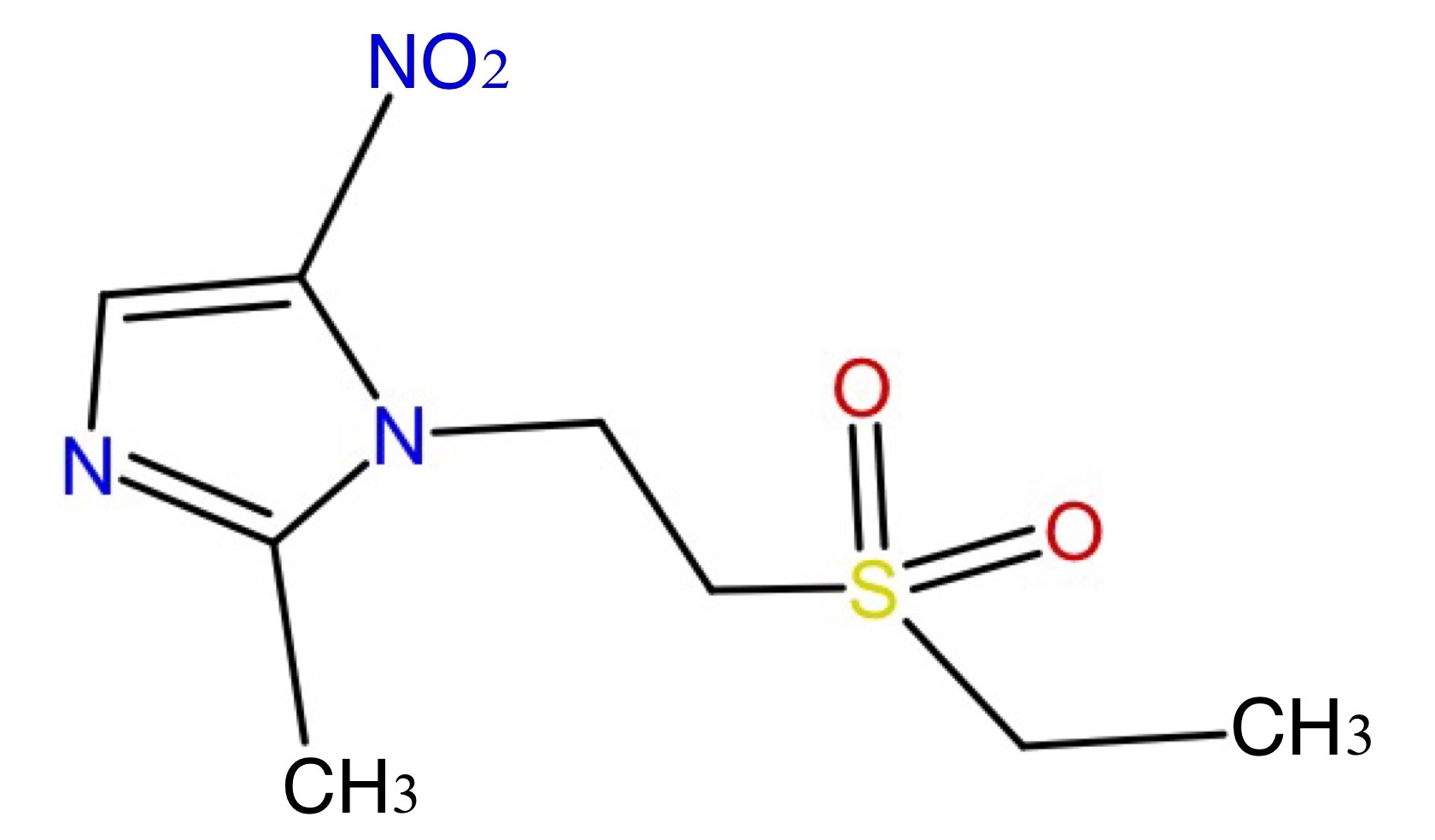
Abb.6: Tinidazol, C8H13N3O4S
Stoffcharakterisierung (Wirkung und Anwendung)
Tinidazol ist ein Wirkstoff aus der Gruppe der Nitroimidazole, der als weißlich bis blassgelbes, kristallines Pulver vorliegt. Die Substanz ist löslich in Aceton und in Dichlormethan, wenig löslich in Methanol und praktisch unlöslich in Wasser. Aufgrund der antiparasitären und bakteriziden Eigenschaften wird Tinidazol für die Behandlung von Protozoonosen, zum Beispiel bei Trichomonas- oder Amöben-Infektionen, und bei Infektionen mit obligat anaeroben Bakterien eingesetzt. Die Wirkung basiert dabei auf einer Nucleinsäuresynthesehemmung.
Eignung der HPLC zur Reinheitsprüfung
Im Arzneibuch ist für Tinidazol unter anderem eine Prüfung auf die verwandten Substanzen 2-Methyl-5-nitro-1H-imidazol (Verunreinigung A) und 1-[2-(Ethylsulfonyl)ethyl]-2-methyl-4-nitro-1H-imidazol (Verunreinigung B) vorgeschrieben. Hierfür eignet sich besonders die HPLC als Analysemethode, da sich die Substanzen mit geeigneter stationärer und mobiler Phase gut trennen lassen und eine Quantifizierung anhand der ⚠ $AUC$ leicht möglich ist. Die Umkehrphasenchromatographie weist hierbei eine gute Trennleistung auf, da sich die Substanzen zwar in polaren Lösungsmitteln etwas lösen (vgl. Stoffcharakterisierung), anderseits aber eine so geringe Polarität aufweisen, dass sie von der hydrophoben stationären Phase retendiert werden können.
Durchführung
Für die Analyse werden vier unterschiedliche Lösungen hergestellt. Für die Untersuchungslösung werden zunächst 10 mg Substanz und für die Referenzlösung b 5 mg der Verunreinigungen A und 5 mg der Verunreinigung B in 10 ml Methanol gelöst und anschließend mit der mobilen Phase verdünnt. Um die Referenzlösungen a und c herzustellen werden die Untersuchungslösung und die Referenzlösung b weiter verdünnt. Die Massenkonzentrationen der Analysesubstanz in der Untersuchungslösung und in Referenz a sind dabei 100 mg/L und 0,1 mg/L. Bei den Lösungen b und c entspricht die Massenkonzentration der Verunreinigungssubstanzen je 10 mg/L und 0,2 mg/L . Die Bestandteile der Lösungen werden mittels HPLC getrennt und anschließend UV/Vis-spektroskopisch bei 320 nm detektiert. Als mobile Phase dient bei dieser Monographie ein Gemisch aus Wasser, Methanol und Acetonitril, die stationäre Phase besteht aus octylsilyliertem Kieselgel. Die Referenzlösung b dient dazu auf eine ausreichende Trennung der Verunreinigungen zu prüfen und eine ausreichende Auflösung sicherzustellen. Die anderen drei Lösungen werden für die Auswertung herangezogen. Es ist darauf zu achten, dass die Lösungen vor Licht geschützt werden, um photolytischen Reaktionen und somit Verunreinigungen aufgrund von Spaltungsprodukten zu verhindern.
Auswertung / Interpretation
Der Grenzwert für die Verunreinigungen A und B liegt bei 0,2 Prozent. Somit sollte die jeweilige Fläche der Peaks bei der Referenzlösung c nicht größer sein als bei der Untersuchungslösung, sonst entspricht die Substanz den Anforderungen des Arzneibuchs nicht. Zusätzlich dürfen die Peakflächen der nicht spezifizierte Verunreinigungen nicht größer als die Fläche des Hauptpeaks der Referenzlösung a sein, sie müssen also weniger als 0,1 Prozent der Probe ausmachen. Die Verunreinigungen dürfen insgesamt nicht mehr als 0,4 Prozent der Analysesubstanz ausmachen.
Einzelnachweise 1 2 3 4 5
1 Ph. Eur. 10.0/1051 Tinidazol; (Eigene Zeichnung; Heteroatome farbig hervorgehoben) ⇑
2 Ph. Eur. 10.0/1051 Tinidazol ⇑
3 https://de.wikipedia.org/wiki/Nitroimidazole (18.05.2021) ⇑
4 Kommentar zu Ph. Eur. 10.0/1051 Tinidazol ⇑
5 Kommentar zu Ph. Eur. 10.0/0675 ⇑
TU-Braunschweig Institut für Medizinische und Pharmazeutische Chemie Seminar: Instrumentelle Analytik Kontakt: tubs@t-kellner.de
