Massenspektrometrie
Bericht der Expertengruppe für
Massenspektrometrie
SoSe 2021
Abgabedatum
21.06.2021
Über-/Expertengruppe 07
Louisa Schönberg
Felicia Schmieding
Michaela Meißner
Marleen Strunk
Lea Ungar
Katharina Morawietz
Massenspektrometrie
Inhaltsverzeichnis
Einleitung
Die Massenspektrometrie ist eine wichtige analytische Methode zur Identifizierung und Strukturaufklärung von chemischen Verbindungen. Sie eignet sich aber ebenfalls für quantitative Bestimmungen.
Bereits Anfang des 19. Jahrhundert stellte William Prout die erste Hypothese zur Massenspektrometrie auf. Er postulierte, dass es eine Eigenschaft des Atoms sei, eine bestimme Masse zu haben.
J. J. Thomson und F. W. Aston entwickelten von 1907-1919 den Massenspektrographen. Hierdurch gelang ihnen letztlich der Nachweis von unterschiedlichen Isotopen eines Elements.
Als Analysenmethode hatte die Massenspektroskopie ihren Durchbruch aber erst um 1960 in der organischen Chemie.
Durch die Massenspektrometrie ist es möglich mit nur wenig Substanz (0,5 mg bis 10 µg) ein Massenspektrum einer Substanz aufzunehmen. Dieses kann dann Auskunft über die relative Molmasse und die Elementarzusammensetzung der Verbindung geben. Bei der Massenspektrometrie kommt es außerdem zum Zerfall von Substanzen in zahlreiche Fragmente, welche weitere Informationen über die Molekülstruktur liefern können. 1
Chemische und physikalische Grundlagen
Grundvorgänge der Massenspektrometrie
Die Massenspektrometrie umfasst verschiedene Grundvorgänge.
Der erste Schritt ist die Ionisation, bei der die Moleküle in einen positiven oder negativen Ladungszustand versetzt werden. Sind diese Molekülionen nicht stabil, kann es zu einer Fragmentierung kommen. Die Moleküle zerfallen schließlich in geladene oder ungeladene Fragmente. Jede Substanz hat dabei seine eigenen, ganz spezifischen Bruchstücke. Diese sind mit einem Fingerabdruck vergleichbar.
Im nächsten Schritt kommt es zu einer Massen-Fokussierung der geladenen Fragmente, wodurch diese nach ihrem Masse-zu-Ladung-Verhältnis aufgetrennt werden.
Zum Schluss werden die Massen und die relativen Häufigkeiten der geladenen Bruchstücke im Massenspektrum registriert.
Die Ionisation kann durch unterschiedliche Methoden erreicht werden, dazu zählen:
- Elektronenstoßionisation
- Chemische Ionisation
- Elektrospray-Ionisation
- Chemische Ionisation unter Atmosphärendruck
- Fast Atom Bombardment
- Matrix-assisted Laser Desorption/Ionisation.
Für die notwendige Massen-Fokussierung kann auf unterschiedliche Methoden zurückgegriffen werden. Dazu zählen die Magnet- und die Elektrostatische Fokussierung. Bei diesen Verfahren werden die Ionen entweder im elektrischen oder im magnetischen Feld beschleunigt und aufgrund ihrer Massen getrennt. Weitere Methoden sind die Quadrupol- und die Flugzeit-Fokussierung.
Um anschließend die Fragmente identifizieren zu können, wird deren Ladung über Ionenauffänger, Photoplatten, Sekundärelektronenvervielfacher oder Ionenfallen analysiert. 2
Masseneinheiten
Für Atom-, Molekül- und Fragmentmassen gilt das Internationale Einheitensystem (SI). Sie werden in der atomaren Masseneinheit „u“ oder „amu“ angegeben. Ersteres steht für unified atomic mass unit, letzteres für atomic mass unit. Das Arzneibuch verwendet die Abkürzung AME. 1 amu entspricht 1,66 ⋅ 10-27 kg. Das entspricht ⚠ $\frac{1}{12}$ der Masse eines Atoms des Kohlenstoffisotops 12C. Daraus ergibt sich die Atommasse des 12C Atoms von 12,0000 amu.
Da sich ein Element aus einer größeren Menge seiner verschiedenen Isotope zusammensetzt, resultiert daraus eine mittlere Atommasse. Diese beträgt beim Kohlenstoff 12,011 amu und beim Wasserstoff 1,00794 amu. Wenn in der Massenspektroskopie Isotope erfasst werden, wird jedes Isotop einzeln und nicht als Mischung registriert. Aus diesem Grund wird für Berechnungen nicht die mittlere Atommasse verwendet, sondern die Masse des reinen Elements.
Ergänzend sei noch die relative Atommasse genannt, welche angibt, wie viel schwerer ein Atom im Vergleich zu ⚠ $\frac{1}{12}$ der Masse des 12C-Atoms ist. Diese ist eine reine Verhältniszahl ohne Einheit. 3
Fragmentierung
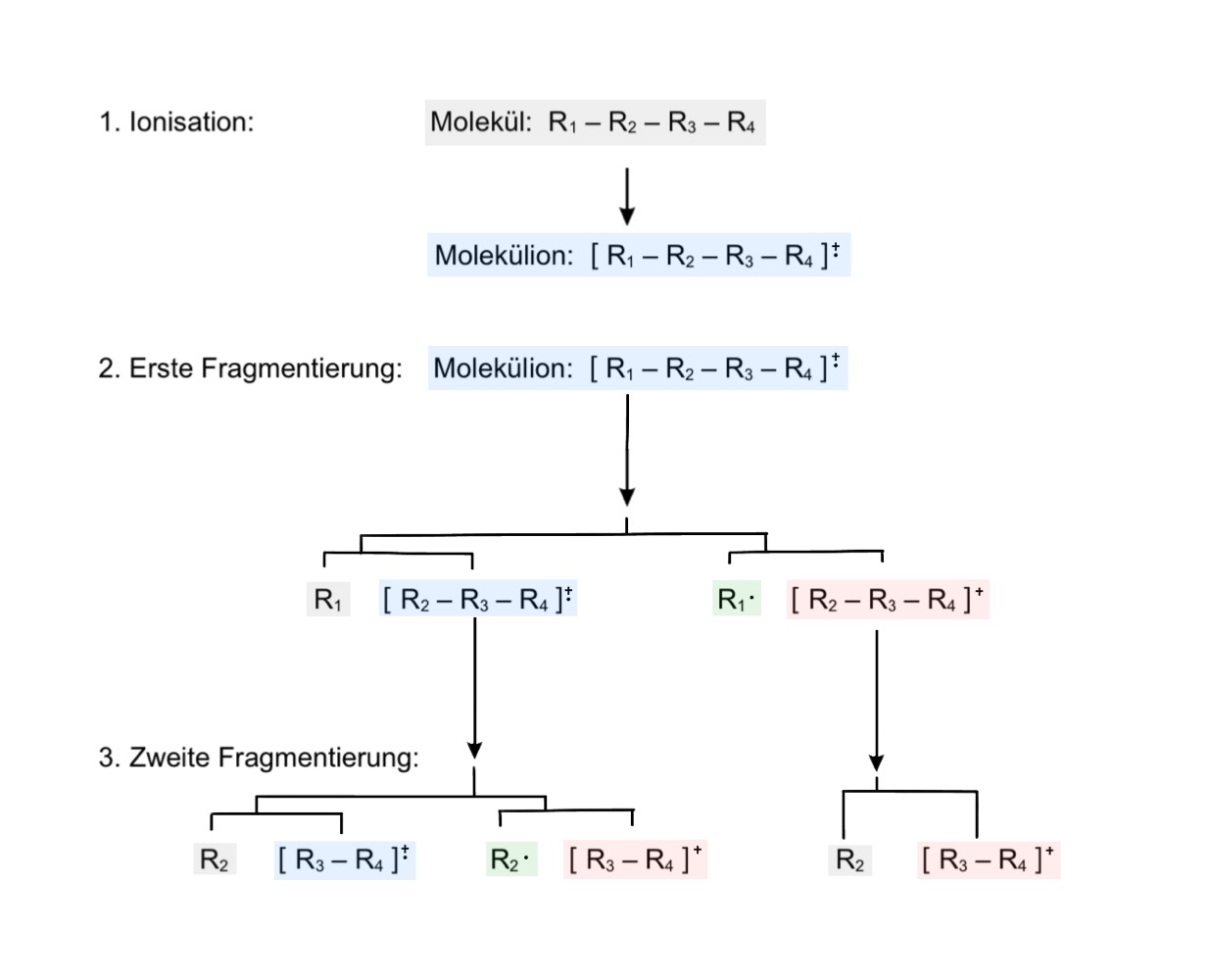
Fragmentierung tritt vor allem bei der harten Ionisation auf (siehe Kapitel 2.2) und ist für die Bildung der spezifischen Spektren der einzelnen Substanzen verantwortlich. Dabei kann sie auch Informationen zum Aufbau der Substanz liefern.
Die Energie, die durch die Ionisation bereitgestellt wird, wird nicht von allen Molekülen und auch oft nicht vollständig aufgenommen. Wenn jedoch die aufgenommene Energie mindestens der Aktivierungsenergie einer Zerfallsreaktion entspricht, kommt es zur Fragmentierung. Alle anderen Moleküle, die durch fehlende aufgenommene Energie nicht fragmentieren, werden später im Molpeak erfasst. Somit gibt das Molpeak Auskunft über das Masse-zu-Ladungs-Verhältnis des intakten Moleküls. Durch die Ionisationsquelle wird das Molekül zunächst ionisiert, wodurch ein radikalisches Molekülkation entstehen kann.
Dieses radikalische Molekülkation kann auf zwei Arten zerfallen:
- durch Abspaltung eines Neutralmoleküls, wobei ein Radikalkation verbleibt
- durch die Abspaltung eines Radikals, wobei ein Kation verbleibt
In der Abbildung sind zur besseren Übersicht alle Radikalkationen blau, Neutralmoleküle grau, Kationen rot und Radikale grün hinterlegt. Die geladenen Fragmente können in einem nächsten Ionisierungsschritt erneut auf die selbe Weise zerfallen. Zu beachten ist hierbei, dass sich ein Kation unter normalen Bedingungen nicht in zwei ungeladene Radikale aufspaltet, da dies energetisch ungünstig ist. 5
Alle Molekülkationen (rot hinterlegt) können später im Detektor erfasst werden und liefern so das Massenspektrum. Radikale und ungeladene Moleküle werden nicht erfasst.
Welche Bindungen des Moleküls gespalten werden hängt von verschiedenen Faktoren ab, u.a.:
- von der Bindungsenergie der jeweiligen Bindungen
- der Stabilität der entstehenden Fragmente
- den Geschwindigkeitskonstanten der einzelnen Zerfallsreaktionen
Dadurch kommt es für die meisten Substanzen zu einer sehr charakteristischen Bindungsspaltung und somit zu einem charakteristischen Massenspektrum. 6
Stephenson-Audier-Regel
Nach der Stephenson-Audier-Regel übernimmt bei einer Bindungsspaltung das Fragment mit dem kleineren Ionisierungspotential die Ladung des Moleküls. Das Ionisierungspotential ist die Energie, die benötigt wird,
um ein Elektron eines Moleküls das sich in Gasphase befindet, heraus zu lösen. 7
Massenspektrum
Ein Massenspektrum ist eine graphische, zweidimensionale Darstellung der detektierten Ionenhäufigkeit im Spektrometer zum Quotienten aus Masse (m) und Ionenladung (z) der nachgewiesenen Ionen. Auf der x-Achse ist das m/z-Verhältnis und auf der y-Achse die Intensität der Signale in Relation zu den relativen Intensitäten der Peaks aufgetragen. Die Skalierung der y-Achse richtet sich dabei nach dem Peak mit der höchsten Signalintensität, dem Basispeak. Dieser liegt bei 100 % der relativen Intensität. Auf die Intensität des Basispeaks bezieht sich die Berechnung der relativen Intensitäten von allen weiteren Peaks.
Die Einheit des Masse-Ladungs-Verhältnisses ist: ⚠ $\tfrac{amu}{e_{0}}$ = 1,04⋅10-8 kg ⋅ C-1. (e0 = 1,6 ⋅ 10-19 Coulomb)
In der Elektronenstoßionisiation-Massenspektroskopie entspricht die Ladung z der Teilchen in der Regel +1.
Dadurch kann die Massenzahl m direkt von der x-Achse abgelesen werden.
Wenn es sich allerdings um Verbindungsklassen wie beispielsweise Aromaten und Heteroaromaten handelt, können auch doppelt geladene Ionen auftreten. Daraufhin wird an der x-Achse m/2 erfasst. Die relative Intensität der Signale von diesen Ionen ist in Folge dessen nur noch halb so groß.
Alternativ zum Basispeak kann die Intensität der Signale von allen positiv entstandenen Ionen auch prozentual angegeben werden. Hierzu wird die Summe aus allen Signalhöhen gebildet. Diese wird dem Totalionenstrom von 100 % gleichgesetzt. Darauf bezogen wird der prozentuale Anteil der einzelnen Signale ermittelt. Dadurch lässt sich ableiten, mit welcher Intensität ein Ion an der Gesamtheit aller positiven Teilchen während des Fragmentierungsprozesses beteiligt ist. 8
Instrumenteller Aufbau
Einlasssysteme
Zum Einführen der Probe in das Vakuumsystem werden bei der Massenspektrometrie Einlasssysteme verwendet. Basierend auf den Eigenschaften der zu analysierenden Substanz kommen verschiedene Einlasssysteme zur Anwendung. Nachfolgend werden zwei Typen von Einlasssystemen beschrieben:
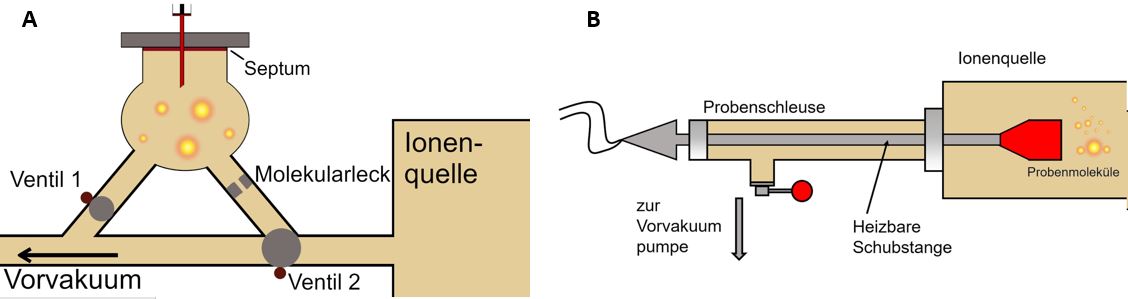
Indirekter Einlass
Bei einem indirekten Einlasssystem wird das Probengefäß vor dem Einbringen der Probe evakuiert. Dies geschieht durch die Öffnung eines ersten Ventils, bedingt durch eine eingeschaltete Vorvakuumpumpe. Dadurch werden alle eventuell im Probengefäß befindlichen Gase aus diesem entfernt. Durch ein Septum hindurch wird mit einer Spritze der flüssige oder gasförmige Analyt in das evakuierte Probengefäß injiziert. Die Temperatur im Inneren des Probengefäßes kann entsprechend der Analyse in einem Temperaturbereich von 20 °C bis maximal 250 °C eingestellt werden. Das zweite Ventil wird geöffnet, damit die Probe in die Ionenquelle gelangt und die Messung vorgenommen werden kann. Abschließend wird das Probengefäß durch erneute Evakuierung von Probenrückständen befreit, wofür Ventil 2 geschlossen vorliegen muss (siehe Abbildung A).
Das Einbringen von leichtflüchtigen oder gasförmigen Substanzen findet häufig über den indirekten Probeneinlass statt, wobei entweder ein Molekularleck (Effusionsdrüse) oder ein Nadelventil verwendet wird. Diese Vorrichtungen dienen der Regulation des Analytanteils, der in die Ionenquelle gelangt, da dieser nicht zu groß sein darf. Das Molekularleck ist eine feine Öffnung, durch welche der Analyt in die Ionenquelle strömen kann. Bei dem Nadelventil hingegen besteht die Möglichkeit, einen konstanten Gasstrom vom Vorratsgefäß zur Ionenquelle einzustellen, da sich das Ventil zwischen den beiden Gerätebauteilen befindet.
Vorteilhaft an einem indirekten Einlasssystem ist, dass so gut wie keine Entmischungsgefahr der Analyse besteht und somit Quantifizierungen gut durchführbar sind. 10 Des Weiteren liefert die Massenspektrometrie unter Verwendung eines solchen Einlasssystems leicht reproduzierbare Spektren. Außerdem besteht die Möglichkeit, bei Verwendung einer Referenzsubstanz diese mit dem Analyten in die Ionenquelle einströmen zu lassen. 11
Nachteile dieses Systems entstehen vor allem durch die Temperatur und Reaktionen mit der Gefäßwand des Geräts, welche zu einer Zersetzung der Probe führen können. Außerdem ist es möglich, dass die zum Aufheizen benötigte zugeführte Energie einen Störfaktor darstellt, der Einfluss auf das Fragmentierungsmuster des Analyten hat. 12
Direkter Einlass
Für die Einführung schwer flüchtiger Substanzen und thermolabiler Verbindungen in das Vakuumsystem werden direkte Einlasssysteme genutzt. In kondensierter Form wird die Probe an einer Schubstange mit speziellem Träger und einem beheizbaren Probengefäß, z.B. einem Tiegel oder einer Drahtschleife an der Spitze in die Probenschleuse eingeführt. Diese wird über eine Vorvakuumpumpe evakuiert. Anschließend wird durch die Öffnung eines Verschlussdeckels an der Ionenquelle die Schubstange in das Hochvakuumsystem eingeschoben (siehe Abbildung direkter Einlass). Das Probengefäß wird elektrisch bis zum Verdampfen der Probe erhitzt, was einen schonenden Prozess aufgrund des geringen Drucks darstellt. Eine Kühlung der Schubstange für leichtflüchtige Stoffe ist auch möglich. Wenn die Verdampfung direkt in das CI-Plasma (induktiv gekoppeltes Plasma) stattfindet, spricht man von einer DEI (direkte Elektronenstoß-Ionisation) oder DCI (direkte chemische Ionisation) (siehe Abbildung B).
Vorteile dieses Einlasssystems sind die Möglichkeit der Temperaturanpassung für unterschiedliche Proben und die dadurch verringerte Gefahr der katalytischen Zersetzung, womit die Trennung einfacher Stoffe und Stoffgemische möglich ist. Nachteile sind die Gefahr der ungleichmäßigen Verdampfung, welche das System für komplexe Stoffgemische ungeeignet macht. Zudem können leichtflüchtige Verunreinigungen mit dem Analyten verwechselt werden. Außerdem können quantitative Analysen nicht durchgeführt werden und die Quelle kann durch große Probenmengen verschmutzt werden, was zu Überlagerungen der nachfolgenden Spektren führen kann.
Unterschiede zum indirekten Einlasssystem sind unter anderem die für den direkten Einlass geringere benötigte Probenmenge, kürzere Wege und die niedrigeren Verdampfungstemperaturen, welche die Zersetzungsrate verringern. 13 14 15
Ionisationsquellen
In der Ionisationsquelle werden die Moleküle der Probe ionisiert. Man unterscheidet zwischen harten und weichen Methoden. Bei der harten Ionisation wird die Probe mit energiereichen Teilchen beschossen, sodass das Molekül ionisiert und stärker fragmentiert wird. Bei der weichen Ionisation können größere Moleküle (z.B. Proteine) angeregt werden. Durch die geringe Energie bleiben die Moleküle intakter und es entstehen größere Fragmente. Es werden bevorzugt Quasimoleküle, wie [M+H] ⁺ /[M-H] ⁻, gebildet. Bei Quasimolekülen unterscheidet sich die Molmasse der ionisierten Probe von der Molmasse der Probe. Die Fragmentierung liefert Informationen zum atomaren Aufbau des Moleküls. 16
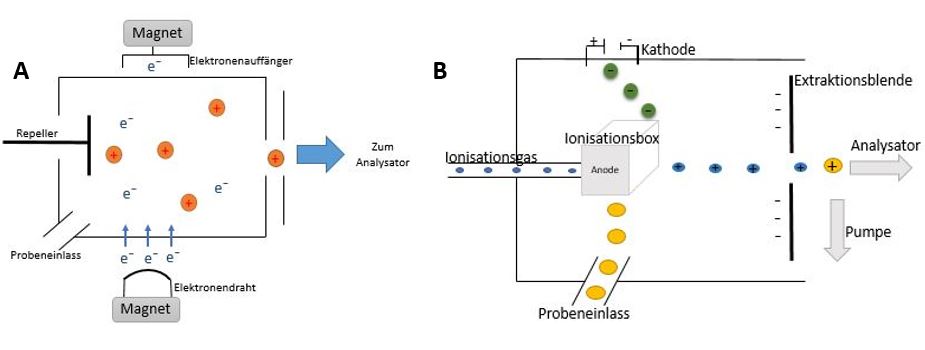
Elektronenstoßionisation (EI)
Bei der Elektronenstoßionisation handelt es sich um eine harte Ionisation, bei der die Moleküle in der Probe mit meist 70 eV geladenen Teilchen beschossen werden. Die Kammerspannung, die zwischen einem beheizten Metalldraht und einem Elektronenauffänger (Anode) anliegt, beschleunigt die geladenen Elektronen. Diese treffen senkrecht auf das Molekül der Probe, lösen Elektronen heraus und ionisieren sie (M + e⁻ → M⁺ + 2e⁻). Die ionisierten Probenbestandteile werden im Anschluss von negativen Beschleunigungselektroden aus der Beschusszone entfernt. Probenbestandteile, die noch nicht ionisiert wurden, werden mit Hilfe eines Vakuums abgesaugt. Durch einen Repeller werden die Ionen beschleunigt, da dieser die geladenen Ionen abstößt und im Anschluss zum Ausgangsspalt und somit zum Analysator befördert (siehe Abbildung A). 18
Chemische Ionisation (CI)
Die chemische Ionisation ist eine weiche Ionisation und ähnelt im Aufbau der EI. Die Unterschiede bestehen darin, dass sich in der CI auch eine Leitung für die Zuführung des Reaktantgases (z.B. Methan, Wasserstoff, Ammoniak) in der Ionisationsbox befindet. Die Wahl des Gases beeinflusst die Intensität der Fragmentierung des Moleküls. Außerdem ist die Kammer gasdicht konstruiert, weshalb sich ein stabiles CI-Plasma ausbilden kann und ein zu schnelles Abpumpen des Reaktantgases verhindert wird. In der Ionisationsbox wird ein Überschuss Reaktantgas zusammen mit der Probe eingeführt und durch einen Elektronenstrahl der Kathode ionisiert. Zuerst wird das Reaktantgas ionisiert und danach entsteht das CI-Plasma. Das gebildete CI-Plasma protoniert die Probe durch Ladungsübertragung. Dabei wird bei Gasen ohne Wasserstoffatom die Ladung direkt übertragen und bei Gasen mit Wasserstoffatomen entstehen Quasimolekülionen. Durch die Extraktionsblende mit Austrittsspalt werden die gebildeten Ionen zum Analysator abtransportiert (siehe Abbildung B). 19
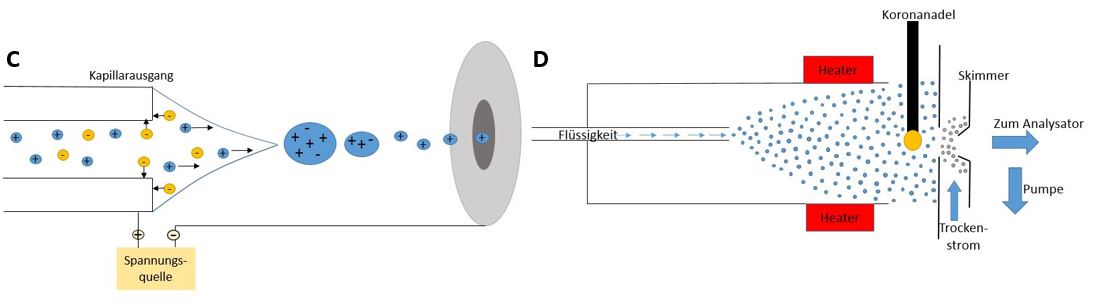
Elektrospray Ionisation (ESI)
Die ESI ist eine weiche Ionisation und eine der am häufigsten angewandten Ionisationsmethoden. Die Probenlösung wird durch eine Stahlkapillare in die Quelle eingebracht. Zwischen der Stahlkapillare und der Gegenelektrode, die sich in der Kammerinnenwand befindet, liegt eine Spannung von 3-6 kV an. Zusätzlich wird die Probe mit einem Inertgas, z.B. Stickstoff, vermischt. Die Probe wird beim Austritt aus der Kapillare vernebelt. Es entsteht ein Aerosol aus Tröpfchen, die je nach Polarität der Hochspannung eine positive oder negative Überschussladung besitzen. Das Lösungsmittel verdampft und die Größe der Tröpfchen verkleinert sich, wobei sich die Oberflächenladung erhöht. Wenn das Verhältnis von Ladung zur Oberfläche einen bestimmten Grenzwert überschreitet, kommt es zu einer Coulomb-Explosion. Dieser Vorgang wiederholt sich, bis die Tröpfchen so klein sind, dass sie nur noch ein Analytmolekül und mehrere Lösungsmittelmoleküle enthalten und einen Radius von 10nm haben. Die Bildung der Tröpfchen wird durch einen Trockengasstrom gefördert. Danach gelangen die so gebildeten Kationen,bzw.Anionen zum Analysator (siehe Abbildung C). 21
Chemische Ionisation unter Atmosphärendruck (APCI)
Die APCI ist eine weiche Ionisationsmethode, bei der die gelöste Probe durch eine beheizte Kapillare gelangt. Dadurch verdampfen unter Atmosphärendruck die Probe und das Lösungsmittel. Das entstandene Aerosol trifft auf eine unter Spannung stehende Koronanadel, die sich zwischen der Kapillare und dem Einlass in das Massenspektrometer befindet. An der Spitze der Nadel entstehen aus dem verdampften Lösungsmittel und aus Luftmolekülen durch Koronaentladung Ionen, welche die Probe ionisieren. Der Protonentransfer ist im positiven Modus (zum Analytmolekül hin) und im negativen Modus (vom Analytmolekül weg) möglich. Die Ionen gelangen durch einen Trockenstrom und durch einen Oberflächenabsauger (Skimmer) in das Hochvakuum und dann zum Analysator (siehe Abbildung D). 22
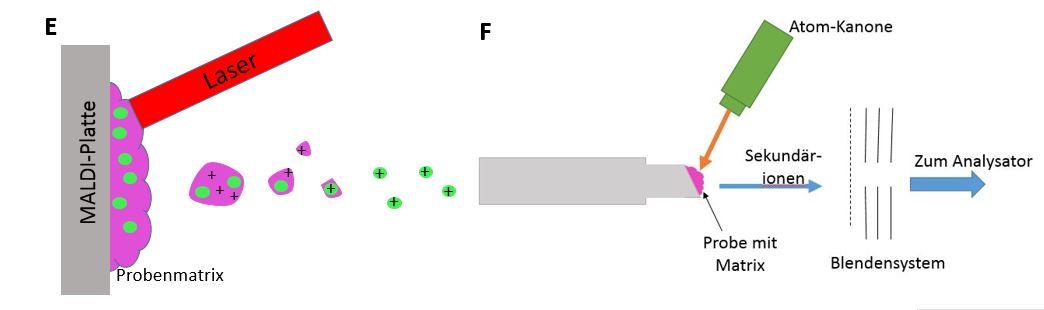
Matrixunterstützte Laserdesorption/-ionisation (MALDI)
Die MALDI ist eine weiche Ionisationsmethode, bei der die Probe in einem niedrig siedenden Lösungsmittel verteilt und anschließend auf einen MALDI-Probenträger aufgebracht wird. Die Matrix wird so ausgewählt, dass sie bei der verwendeten Wellenlänge ein Absorptionsmaximum besitzt. Auf dem Träger entsteht durch Trocknung ein Kristallfilm. Dieser wird mit einem Laserstrahl (Stickstofflaser) beschossen und somit ionisiert und in die Gasphase überführt. Es handelt sich um eine Protonenübertragung von der Matrix auf die Probe, wodurch Quasimoleküle entstehen. Danach werden die Matrix- und Probenmoleküle detektiert und ins Vakuum desorbiert. Diese Methode findet nur Anwendung in Kombination mit dem Flugzeit-Analysator (siehe Massenanalysatoren) (siehe Abbildung E). 24
Fast Atom Bombardment (FAB)
Bei dem FAB handelt es sich um eine weiche Ionisation für schwere und nichtflüchtige Substanzen. Diese werden zusammen mit einer im Überschuss vorliegenden, schwerflüchtigen Matrix (meist Glycerin), auf einen Träger aufgebracht. Der Träger wird mit einer Atom-Kanone (energiereiche Atome: Xe, Ar) beschossen und es entstehen Quasimolekülionen und Glycerin-Cluster. Die so gebildeten Sekundärionen werden durch ein elektrisches Feld extrahiert und durch ein Blendensystem zum Analysator beschleunigt (siehe Abbildung F). 25
Massenanalysatoren
Massenanalysatoren selektieren den Ionenstrahl nach ihrem Masse-zu-Ladung-Verhältnis (m/z-Verhältnis). Außerdem stellen sie sicher, dass alle Ionen die in den Detektor gelangen die gleiche Richtung und Geschwindigkeit aufweisen, um somit fehlerfrei detektiert werden zu können.
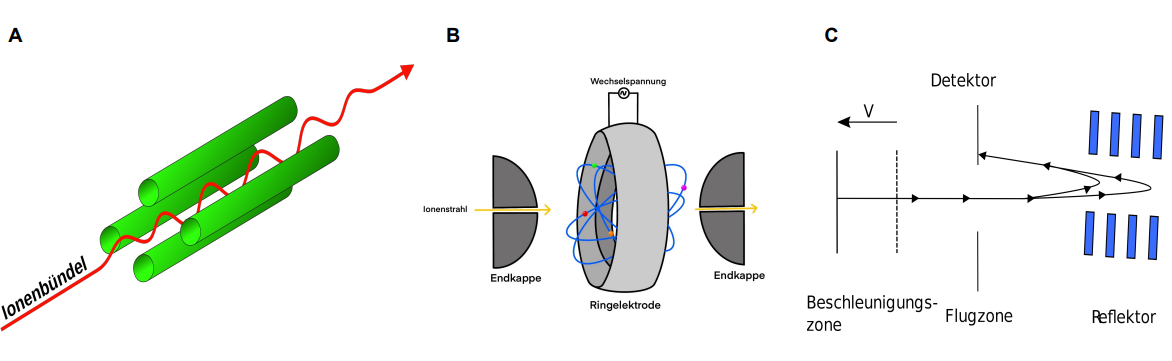
Sektorfeld Analysator
Der Sektorfeld-Analysator ist aus einem stumpfwinklig gebogenen Metallrohr aufgebaut, dessen Winkel z.B. 60°, 90°, 120° oder 180° betragen kann. Das Metallrohr wird so zwischen einen Elektromagneten gebracht, dass das erzeugte Magnetfeld senkrecht zum Ionenstrahl wirkt. Die Stärke des Magnetfeldes, also die magnetische Flussdichte B, kann durch die Änderung der Spulenstromstärke variiert werden. Die geladenen Teilchen, die mit dem Ionenstrom in den Analysator gelangen, werden in dem Magnetfeld durch die Lorentz-Kraft abgelenkt. Dabei wirkt die Lorentz-Kraft senkrecht zur Bewegungsrichtung der Ionen und senkrecht zum Magnetfeld. Daraus resultiert eine kreisförmige Ablenkung der Ionen.
Für die Lorentz-Kraft ⚠ $ F_L $ gilt:
⚠ $ F_L = B \cdot z \cdot v $
⚠ $ B $ = magnetische Flussdichte⚠ $ v $ = Geschwindigkeit der Teilchen⚠ $ z $ = Ladung der Teilchen
Aus der Formel ergibt sich, dass die Lorentz-Kraft direkt abhängig von der Geschwindigkeit ⚠ $ v $ der Teilchen, sowie ihrer Ladung ist und zusätzlich durch das anliegende Magnetfeld beeinflusst wird.
Da die Geschwindigkeit eines Teilchens unmittelbar von der Masse abhängt,
kann man durch das Anlegen eines konstanten Magnetfeldes, die Ionen nach einem bestimmten m/Z-Verhältnis filtern.
Gleichzeitig wirkt auf die Ionen, die sich auf einer Kreisbahn befinden, die Zentrifugalkraft entgegen der Lorentz-Kraft. Die beiden Kräfte gleichen sich aus, sodass die Ionen quasi kräftefrei fliegen.
Für die Zentrifugalkraft ⚠ $ F_z $ gilt:
⚠ $ F_z = \frac{m \cdot v^{2}}{r} $ ⚠ $\qquad$ ⚠ $v=\sqrt{\frac{2z \cdot U}{m}}$
⚠ $ U $ = Beschleunigungsspannung⚠ $ m $ = Masse des Teilchens⚠ $ r $ = Radius der Kreisbahn
Indem man nun das Magnetfeld kontinuierlich ändert, kann man nacheinander Ionen mit verschiedene m/z-Verhältnissen zum Detektor gelangen lassen und so ein Massenspektrum erstellen. Alle anderen Ionen die nicht dem passendem m/z-Verhältnis entsprechen, werden zu stark/wenig abgelenkt, treffen auf die Wände des Analysators und gelangen somit nicht zum Detektor.
Alternativ kann auch die Beschleunigungsspannung verändert und das magnetische Feld konstant gehalten werden, dies wird allerdings seltener angewandt.
Aus ⚠ $ F_z = F_L $ ergibt sich durch Umstellen die Grundgleichung der Magnet-Fokussierung, mit der man das m/z-Verhältnis berechnen kann: 29
⚠ $ \frac{m}{z} = \frac{r^2 \cdot B^2}{2 \cdot U} $
Quadrupol-Analysator (Q)
Ein Quadrupol-Massenspektrometer besitzt ein elektrisches Quadrupol als Analysator, welches zur Ionentrennung eingesetzt wird.
Er wird für einen kleinen Massenbereich von bis zu 4000 m/z verwendet.
Dieser Massenspektrometertyp bietet viele Vorteile, da er kompakt, kostengünstig und leicht bedienbar ist. Außerdem hat der Analysator eine kurze Scanzeit, wodurch auch eine Kopplung des Analysators mit chromatographischen Methoden möglich ist.
Der Analysator besteht aus vier parallel zueinander liegenden Metallstäben, welche paarweise als Elektroden dienen (siehe Abbildung A). Die gegenüberliegenden Stäbe werden dabei elektrisch leitend miteinander verbunden. Je eins der gegenüberliegenden Stäbchenpaare wird an den positiven Pol, das andere an den negativen Pol einer Gleichspannungsquelle angeschlossen. Die Gleichspannung wird zusätzlich durch eine hochfrequente Wechselspannung überlagert.
Durch diese Konstruktion erhalten die Stäbe, welche nebeneinander angeordnet sind, eine entgegengesetzte Polarität und eine Wechselspannung mit einer um 180° versetzten Phase.
Im Innenraum entsteht auf diese Weise zwischen den vier Metallstäbchen ein elektrisches Feld. Nur Ionen mit einem bestimmten m/z-Verhältnis können in einer stabilen oszillierenden Flugbahn in Längsrichtung der Stäbchen zum nachgeschalteten Detektor gelangen.
Die Ionen, welche ein anderes m/z-Verhältnis aufweisen, prallen auf die Stäbchen. Dabei werden diese Ionen entladen und der Detektion mit Hilfe einer Vakuumpumpe entzogen.
Für die Erzeugung eines Massenspektrums wird ein Massenbereich gescannt. Dies kann auf zwei unterschiedlichen Wegen erfolgen.
Bei der ersten Methode müssen die Gleichspannung und die Amplitude des Wechselfelds unter Einhaltung des U/V-Verhältnisses bei konstanter Frequenz erhöht werden. Auf diese Weise können die Ionen mit dem nächst höheren m/z-Verhältnis den Detektor erreichen. Die m/z-Werte der Ionen sind dabei proportional zur Spannung.
Bei der zweiten Methode wird die Frequenz erhöht und die Gleich- und Wechselspannung werden konstant gehalten. 30
Ionenfalle
Der Ionenfallen-Analysator ähnelt von der Funktionsweise dem Quadrupolstab, basiert allerdings darauf, dass Ionen mit bestimmten m/z-Verhältnissen auf eine stabile Flugbahn gelenkt und somit in einem System festgehalten werden.
Er ist aus einer Ringelektrode und zwei hyperbolischen Endkappen aufgebaut. Die beiden Endkappen besitzen zentrische Öffnungen, um den Ionenstrahl hinein oder heraus zu lassen. Der Aufbau ist in Abbildung B der Analysatoren dargestellt. An der Ringelektrode wird eine hochfrequente Wechselspannung angelegt, wodurch ein elektrisches Feld im Inneren des Analysators entsteht. Dieses elektrische Feld zwingt die Ionen auf eine elipsoide Flugbahn, die sogenannte „Lissajous-Bahnen“, dadurch verbleiben sie im Inneren des Analysators und sind somit gefangen. Die Bahnen beispielhafter Ionen sind in der Abbildung in blau dargestellt.
In dem Analysator befindet sich Helium-Gas bei einem Druck von 10-4 bis 10-6 bar. Das Helium-Gas sorgt dafür, dass die Ionen abgebremst werden, da das elektrische Feld alleine nicht stark genug ist. 31
Um nun Ionen nach ihrem m/z-Verhältnis zu selektieren, wird meistens eine weitere Wechselspannung an die Endkappen angelegt. Dieser Aufbau wird als „Paul-Falle“ nach dem Physiker Wolfgang Paul bezeichnet. So entsteht im Inneren ein elektrisches Quadrupolfeld (auch Multipolfelder genannt), dass auf die Ionen eine zeitlich periodisch wechselnde Kraft ausübt. Durch die Variation der elektrischen Felder, können entweder nur Ionen mit einem bestimmten m/z-Verhältnis gefangen gehalten werden, oder nur Ionen mit bestimmten m/z-Verhältnis herausgelassen werden und zum Detektor gelangen. 32
Alternativ kann zur Selektion auch die Spannung der Ringelektrode schrittweise erhöht werden, ohne das eine zweite Wechselspannung angelegt wird. Dies wird heutzutage allerdings seltener angewendet, da so nur relativ langsame Scangeschwindigkeiten erreicht werden können.
Die Vorteile der (Quadrupol-)Ionenfalle sind, dass sie eine hohe Nachweisempfindlichkeit besitz und das man Fragmentierungsspektren gezielter Ionen aufnehmen kann, indem man andere Ionen entfernt. Außerdem sind Wiederholungen der Aufnahme eines Massenspektrum in einem Durchlauf möglich. 33
Flugzeit-Analysator (TOF)
Die Aufgabe des Flugzeit-Massenanalysators ist es, Ionen anhand ihrer verschiedenen Flugzeiten zu trennen. Abhängig von der Ionenmasse können sich die Ionen schneller bzw. langsamer bewegen. Leichte Ionen haben eine kürzere, schwerere Ionen eine längere Flugzeit zum Detektor. Der Analysator kann die Flugzeit messen und daraus die Masse des Ions bestimmen.
Ein hierfür typisches Massenspektrometer in der Praxis ist die Kombination aus MALDI und Flugzeit-Analysator. Dabei werden die Ionen in gepulster Form dem Analysator geliefert.
In der Ionenquelle werden die unterschiedlich schweren Ionen gebildet. Durch einen Spannungsimpuls von 103 bis 104 V erhalten alle Ionen dieselbe kinetische Energie. Im Anschluss legen die Ionen innerhalb von maximal hundert Mikrosekunden im TOF-Massenspektrometer eine ein Meter lange Flugstrecke durch ein lineares Flugrohr zum Detektor zurück. Der Detektor registriert das Massenspektrum, erst von den leichten, dann von den schweren Ionen.
Des Weiteren gibt es auch Massenspektrometer mit einem Reflektor-Flugrohr. Dazu wird ein Reflektor zwischen dem Ionen-Eintritt und der Ionen-Detektion eingebaut (siehe Abbildung C).
Durch den Reflektor entsteht ein elektrisches Gegenfeld, wodurch die Ionen auf ihrer Flugbahn ihre Richtung ändern und so zum Detektor gelangen.
Kommt es trotz gleicher Ladung und Masse der Ionen zu Energieunterschieden, dann können diese durch den Reflektor kompensiert werden. Die Ionen mit einer höheren Energie, und somit mit einer schnelleren Geschwindigkeit, können tiefer in den Feldbereich des Reflektors eindringen. Diese Ionen haben im Vergleich zu den langsameren Ionen, bei gleicher Masse, einen längeren Flugweg. Dadurch wird der Geschwindigkeitsunterschied zwischen den langsamen und schnellen Ionen kompensiert.
Im Vergleich zum linearen Flugrohr ist der Flugweg beim Reflektor-Flugrohr doppelt so lang, wodurch eine höhere Auflösung durch Peakschärfung erreicht wird. 34
Zur Herleitung der Flugzeit bedient man sich folgender Formel:
⚠ $t=\sqrt{\frac{s^{2}}{2\cdot E_{kin}}\cdot m}=k\cdot\sqrt{m}$ ⚠ $\qquad$ ⚠ ${E}_{kin}= \tfrac{1}{2}mv^2$
⚠ $ v $ = Geschwindigkeit⚠ $ s $ = Flugstrecke⚠ $ t $ = Flugzeit⚠ $ k $ = Faktor⚠ $ m $ = Masse
Detektoren
Der letzte Bestandteil des instrumentellen Aufbaus eines Massenspektrometers ist der Detektor. Dieser registriert die Intensität, der im Massenanalysator getrennten und in der Ionenquelle gebildeten Ionen und erzeugt ein elektrisches Signal, welches zur Auswertung der Messung genutzt wird. Es gibt viele unterschiedliche Detektoren, die in ihrer Genauigkeit, Empfindlichkeit, Ansprechzeit und ihrem Detektionsbereich variieren. Deshalb wird für jede Messung entsprechend ein bestimmter Detektor ausgewählt, je nachdem, welche Ansprüche an die Messdaten gestellt werden. 35
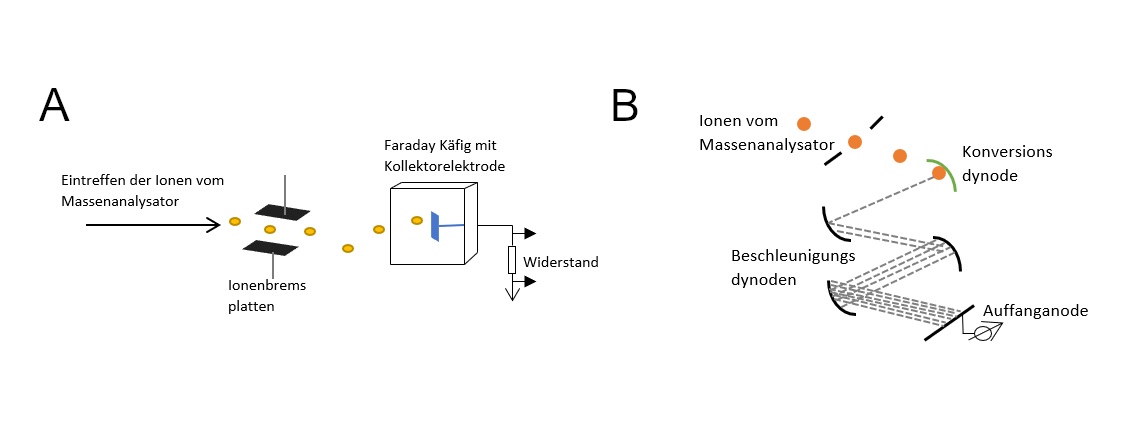
Faraday Auffänger
Der Faraday Auffänger (Abbildung A), der auch Faraday Becher oder Faraday Käfig genannt wird, besteht aus einer Kollektorelektrode, welche von einem Käfig umgeben ist. Dieser sorgt dafür, dass reflektierte Ionen und herausgeschlagene Sekundärelektronen daran gehindert werden aus dem System herauszutreten.
Die einfallenden Ionen werden durch Ionenbremsplatten vor Eintritt in den Käfig auf eine einheitliche Geschwindigkeit abgebremst. Anschließend treffen sie auf die Kollektorelektrode, die so in dem Käfig ausgerichtet ist, dass die Ionen vom Eingang des Bechers weg reflektiert werden sobald sie eintreten. Da die Kollektorelektrode und der Käfig über einen hohen Widerstand mit dem Erdpotential verbunden sind, wird die Ladung der positiven Ionen beim Auftreffen durch den Elektronenfluss der Erde über den Widerstand neutralisiert. Es kommt zu einem Spannungsabfall, der durch einen hochohmigen Verstärker verstärkt wird, sodass ein elektrisches Signal entsteht, welches zur Auswertung herangezogen wird. 37 38
Die Vorteile dieses Detektors sind, dass er preiswert in der Anschaffung ist, über einen einfachen und robusten Aufbau verfügt und eine hohe Genauigkeit hat, da eine Proportionalität zwischen der Intensität der Entladung der Ionen und dem daraus resultierenden Spannungsabfall vorliegt. Außerdem ist der Faraday Auffänger vielseitig einsetzbar, da sein Ansprechverhalten unabhängig von der Energie, Masse oder den chemischen Eigenschaften der Ionen ist. Aufgrund des Anbringens eines hochohmigen Widerstandes, erfolgt die Messung jedoch vergleichsweise langsam und mit einer nicht so hohen Empfindlichkeit, wie es zum Beispiel beim Sekundärelektronenvervielfacher der Fall ist. 39 40 41
Sekundärelektronenvervielfacher (SEV)
Der Sekundärelektronenvervielfacher (Abbildung B) macht sich die Entstehung von sogenannten Sekundärelektronen zu nutzen. Diese Teilchen entstehen beim Auftreffen eines Elektrons auf einer schlecht leitenden Oberfläche. So können aus einem Elektron viele Elektronen mit geringerer Ladung entstehen.
Für den SEV werden Dynoden mit einer Cu/Be-Oberfläche verwendet, an die eine Spannung von bis zu 25 kV angelegt wird. Die Ionen aus dem Massenanalysator treffen auf die erste Dynode, die sogenannte Konversionsdynode, die aus den auftreffenden Ionen Elektronen erzeugt. Sie besitzt eine, den Ionen entgegengesetzte Polarität und ein sehr hohes elektrisches Potential. Die entstandenen Elektronen werden von der Konversionsdynode zur nächsten Dynode hin beschleunigt, wo sie Sekundärelektronen erzeugen. Dieser Vorgang kann über 20 Beschleunigungsdynoden wiederholt werden, sodass eine Signalkaskade entsteht, die an der Auffanganode als elektrisches Signal detektiert wird. 42
Der Sekundärelektronenvervielfältiger hat eine sehr kurze Ansprechzeit von nur wenigen Nanosekunden und durch den Ablauf über mehrere Dynoden bzw. die kontinuierliche Sekundärelektronenerzeugung einen hohen Verstärkungsfaktor und damit eine hohe Genauigkeit, weshalb er oft Verwendung findet. Durch das Abnutzen der Dynoden haben SEV eine kurze Lebensdauer und einen über die Zeit ansteigenden Empfindlichkeitsverlust. Außerdem ist dieser Detektor weniger empfindlich schwereren Ionen gegenüber. 43 44
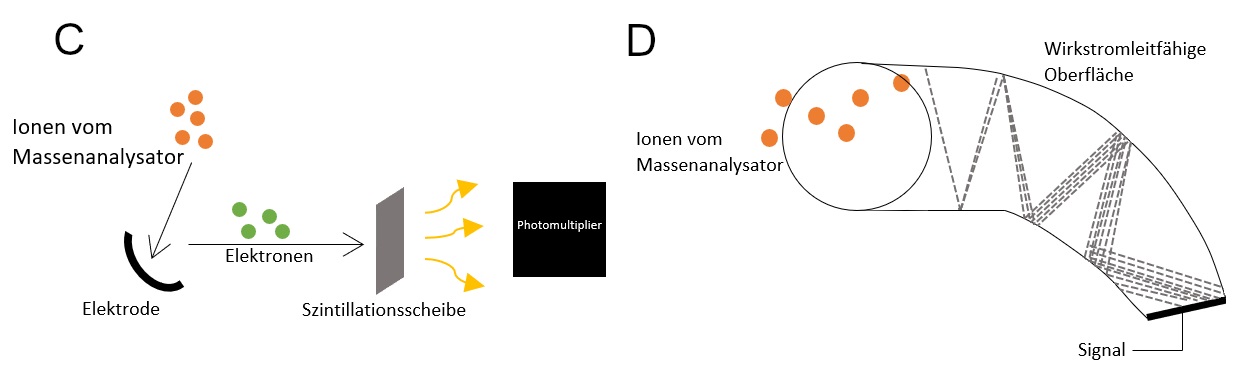
Szintillationsdetektor
Dieser elektrooptische Detektor (Abbildung C) erzeugt aus den eintreffenden Ionen Elektronen, welche anschließend Photonen freisetzen. Die Ionen treffen dabei auf eine Elektrode mit stark negativem Potential, wodurch sie Elektronen herauslösen. Die so entstandenen Elektronen werden im nächsten Schritt zur Szintillationsscheibe hin beschleunigt. Dort erzeugen sie Photonen, welche im Photonenmultiplier verstärkt und schließlich als elektrisches Signal detektiert werden. 46
Der Szintillationsdtektor besitzt eine hohe Empfindlichkeit und eine schnelle Arbeitszeit. Außerdem hat er eine lange Lebenszeit und produziert nur ein geringes Rauschen. Die Anschaffung eines solchen Detektors ist jedoch teuer. 47
Kanalelektronenvervielfacher (KEV)
Dieser Detektor (Abbildung D) besteht aus einem Glasrohr mit einer leitfähigen Innenbeschichtung. Das Prinzip des Kanalelektronenvervielfachers kann mit dem des Sekundärelektronenvervielfachers verglichen werden.
Hier treffen die Ionen aus dem Massenanalysator zunächst auf eine Konversionsdynode aus Halbleitermaterial, aus der in Folge Elektronen herausgelöst werden. Die Elektronen treten dann in das Rohr ein, in dem mehrere parallel geschaltete Kanäle vorhanden sind. An der Innenwand wird ein Spannungsgefälle angelegt, wodurch auch hier eine fortlaufende Elektrodenkaskade durch Sekundärelektronen ausgelöst wird. Diese Kaskade erzeugt ein elektrisches Signal.
In der Massenspektrometrie wird der Kanalelektronenvervielfacher eher weniger eingesetzt, dafür findet er aber Verwendung in der Raman- und UV/VIS-Spektroskopie. 48
Kopplungen
Die Massenspektrometrie wird oft auch mit anderen Analyseverfahren gekoppelt, was vor allem durch die Entwicklung der unterschiedlichen Ionisationsverfahren möglich geworden ist. Eine Kopplung mit Trennverfahren, wie der Chromatographie oder Elektrophorese, bietet große Vorteile in der Analyse von Substanzgemischen. Mögliche Kopplungsarten sind hierbei:
- Die Flüssigchromatographie mit der Massenspektrometrie (LC/MS oder HPLC-MS)
- Die Gaschromatographie mit der Massenspektrometrie (GC-MS)
- Die Thermoanalyse mit der Massenspektrometrie (TA-MS)
- Die Tandem-Massenspektrometrie (MS/MS oder CID)
- Die Kapillarelektrophorese mit der Massenspektrometrie (CE-MS)
Einzelnachweise
1 https://www.chemie-schule.de/KnowHow/Massenspektrometrie, abgerufen: 16.05.2021, 23:15 Uhr ⇑
2 Rücker/Neugebauer/Willems (2013): Instrumentelle pharmazeutische Analytik (5. Auflage), Wissenschaftliche Verlagsgesellschaft Stuttgart ⇑
3 Rücker/Neugebauer/Willems (2013): Instrumentelle pharmazeutische Analytik (5. Auflage), Wissenschaftliche Verlagsgesellschaft Stuttgart ⇑
4 Eigenes Design: L. Ungar ⇑
5 Rücker/Neugebauer/Willems (2013): Instrumentelle pharmazeutische Analytik (5. Auflage), Wissenschaftliche Verlagsgesellschaft Stuttgart ⇑
6 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/masse/ms_ionenquelle_ei.vlu/Page/vsc/de/ch/3/anc/masse/2_massenspektrometer/2_3_ionenquelle/2_3_1_elek_stoss/2_3_1_3_fragmentierung/fragmentierung_910ht0301.vscml.html, abgerufen: 12.05.2021, 12:08 Uhr ⇑
7 Rücker/Neugebauer/Willems (2013): Instrumentelle pharmazeutische Analytik (5. Auflage), Wissenschaftliche Verlagsgesellschaft Stuttgart ⇑
8 Rücker/Neugebauer/Willems (2013): Instrumentelle pharmazeutische Analytik (5. Auflage), Wissenschaftliche Verlagsgesellschaft Stuttgart ⇑
9 Eigenes Design: L. Schönberg ⇑
10 http://www.chemgapedia.de/vsengine/tra/vsc/de/ch/3/anc/massenspektrometrie1.tra/Vlu/vsc/de/ch/3/anc/masse/ms_einlass_vakuum.vlu/Page/vsc/de/c3/3/anc/masse/2_massenspektrometer/2_1_einlasssystem/2_1_1_indirekteinlass/indirekt_m39ht0202.vscml.html, abgerufen: 11.05.2021, 15:55 Uhr ⇑
11 Skript: Seminar zum Praktikum Instrumentelle Analytik SoSe21; Kapitel: Einführung in die Massenspektrometrie von Finja Krebs ⇑
12 Massenspektrometrie von Herbert Budzikiewicz, Mathias Schäfer; Verlag: WILEY-VCH, S.10; 6., vollständig überarbeitete und aktualisierte Auflage ⇑
13 Massenspektrometrie von Herbert Budzikiewicz, Mathias Schäfer; Verlag: WILEY-VCH, S.10; 6., vollständig überarbeitete und aktualisierte Auflage ⇑
14 http://www.chemgapedia.de/vsengine/tra/vsc/de/ch/3/anc/massenspektrometrie1.tra/Vlu/vsc/de/ch/3/anc/masse/ms_einlass_vakuum.vlu/Page/vsc/de/ch/3/anc/masse/2_massenspektrometer/2_1_einlasssystem/2_1_2_direkteinlass/direkt_m39ht0202.vscml.html, abgerufen: 11.05.2021, 20:12 Uhr ⇑
15 Skript: Seminar zum Praktikum Instrumentelle Analytik SoSe21; Kapitel: Einführung in die Massenspektrometrie von Finja Krebs ⇑
16 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/masse/ms_ionenquelle.vlu.html, abgerufen: 12.05.2021, 11:34 Uhr ⇑
17 Eigenes Design M.Strunk ⇑
18 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/masse/ms_ionenquelle_ei.vlu.html, abgerufen: 13.05.2021, 16:12 Uhr ⇑
19 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/masse/ms_ionenquelle_ci.vlu/Page/vsc/de/ch/3/anc/masse/2_massenspektrometer/2_3_ionenquelle/2_3_2_chem_ionisation/2_3_2_1_aufbau_ci/aufbau_22ht0801.vscml.html, abgerufen: 13.05.2021, 18:25 Uhr ⇑
20 Eigenes Design M.Strunk ⇑
21 Rücker/Neugebauer/Willems (2013): Instrumentelle pharmazeutische Analytik (5. Auflage), Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 371-372 ⇑
22 Skript: Seminar zum Praktikum Instrumentelle Analytik SS2021, Kapitel: Einführung Massenspektrometrie von Finja Krebs ⇑
23 Eigenes Design M.Strunk ⇑
24 Skript: Seminar zum Praktikum Instrumentelle Analytik SS2021, Kapitel: Einführung Massenspektrometrie von Finja Krebs ⇑
25 Skript: Seminar zum Praktikum Instrumentelle Analytik SS2021, Kapitel: Einführung Massenspektrometrie von Finja Krebs ⇑
26 https://de.wikipedia.org/wiki/Quadrupol-Massenspektrometer, abgerufen: 16.Mai 2021, 23:25 Uhr ⇑
27 Eigenes Design: L. Ungar ⇑
28 https://de.wikipedia.org/wiki/Flugzeitmassenspektrometer, abgerufen: 16.Mai 2021, 23:45 Uhr ⇑
29 Rücker/Neugebauer/Willems (2013): Instrumentelle pharmazeutische Analytik (5. Auflage), Wissenschaftliche Verlagsgesellschaft Stuttgart ⇑
30 Rücker/Neugebauer/Willems (2013): Instrumentelle pharmazeutische Analytik (5. Auflage), Wissenschaftliche Verlagsgesellschaft Stuttgart ⇑
31 Rücker/Neugebauer/Willems (2013): Instrumentelle pharmazeutische Analytik (5. Auflage), Wissenschaftliche Verlagsgesellschaft Stuttgart ⇑
32 https://de.wikipedia.org/wiki/Paul-Falle, abgerufen: 14.05.2021, 15:53 Uhr ⇑
33 https://www.chemie.de/lexikon/Ionenfallen-Massenspektrometer.html, abgerufen: 14.05.2021, 17:14 Uhr ⇑
34 Rücker/Neugebauer/Willems (2013): Instrumentelle pharmazeutische Analytik (5. Auflage), Wissenschaftliche Verlagsgesellschaft Stuttgart ⇑
35 Massenspektrometrie, die Detektoren: http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/masse/ms_detektor_datensystem.vlu.html, abgerufen: 12.05.2021, 12:40 Uhr ⇑
36 Eigenes Design: M. Meißner ⇑
37 Der Faraday Auffänger: http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/masse/ms_detektor_datensystem.vlu/Page/vsc/de/ch/3/anc/masse/2_massenspektrometer/2_5_detektor_ms/2_5_3_faradaybecher/faraday_ms4ht0202.vscml.html, abgerufen: 12.05.2021, 12:38 Uhr ⇑
38 Skript: Seminar zum Praktikum Instrumentelle Analytik SoSe21; Kapitel: Einführung in die Massenspektrometrie von Finja Krebs ⇑
39 Faraday Becher: https://de.wikipedia.org/wiki/Faraday-Becher, abgerufen: 12.05.2021, 12:38 Uhr ⇑
40 Der Faraday Auffänger: http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/masse/ms_detektor_datensystem.vlu/Page/vsc/de/ch/3/anc/masse/2_massenspektrometer/2_5_detektor_ms/2_5_3_faradaybecher/faraday_ms4ht0202.vscml.html, abgerufen: 12.05.2021, 12:38 Uhr ⇑
41 Instrumentelle Analytik: Grundlagen-Geräte-Anwendungen, Skoog/ Leary, 1996 ⇑
42 Skript: Seminar zum Praktikum Instrumentelle Analytik SoSe21; Kapitel: Einführung in die Massenspektrometrie von Finja Krebs ⇑
43 Der Sekundärelektronenvervielfacher: http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/masse/ms_detektor_datensystem.vlu/Page/vsc/de/ch/3/anc/masse/2_massenspektrometer/2_5_detektor_ms/2_5_5_sev/sev_ms4ht0202.vscml.html, abgerufen: 12.05.2021, 12:37 Uhr ⇑
44 Detektor: https://www.pfeiffer-vacuum.com/de/know-how/massenspektrometer-und-restgasanalyse/quadrupol-massenspektrometer-qms/detektor/, abgerufen: 10.05.2021, 17:12 Uhr ⇑
45 Eigenes Design: M. Meißner ⇑
46 Skript: Seminar zum Praktikum Instrumentelle Analytik SoSe21; Kapitel: Einführung in die Massenspektrometrie von Finja Krebs ⇑
47 Szintillationszähler: https://de.wikipedia.org/wiki/Szintillationsz%C3%A4hler, abgerufen: 12.05.2021, 12:36 Uhr ⇑
48 Der Sekundärelektronenvervielfacher: http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/masse/ms_detektor_datensystem.vlu/Page/vsc/de/ch/3/anc/masse/2_massenspektrometer/2_5_detektor_ms/2_5_5_sev/sev_ms4ht0202.vscml.html, abgerufen: 14.05.2021, 12:32 Uhr ⇑
49 Kopplungsmethoden: https://de.wikipedia.org/wiki/Fl%C3%BCssigchromatographie_mit_Massenspektrometrie-Kopplung, abgerufen: 21.06.2021, 12:03 Uhr ⇑
50 Kopplungsmethoden in der Massenspektrometrie: https://analyticalscience.wiley.com/do/10.1002/gitfach.18009, abgerufen: 10.05.2021, 12:26 Uhr ⇑
51 Skript: Seminar zum Praktikum Instrumentelle Analytik SoSe21; Kapitel: Einführung in die Massenspektrometrie von Finja Krebs ⇑
52 Skript: Seminar zum Praktikum Instrumentelle Analytik SoSe21; Kapitel: Einführung in die Massenspektrometrie von Finja Krebs ⇑
53 Skript: Seminar zum Praktikum Instrumentelle Analytik SoSe21; Kapitel: Einführung in die Massenspektrometrie von Finja Krebs ⇑
Monographiebeispiel: Kupfertetramibitetrafluoroborat, Prüfung auf Gehalt
Stoffcharakterisierung (Wirkung und Anwendung)
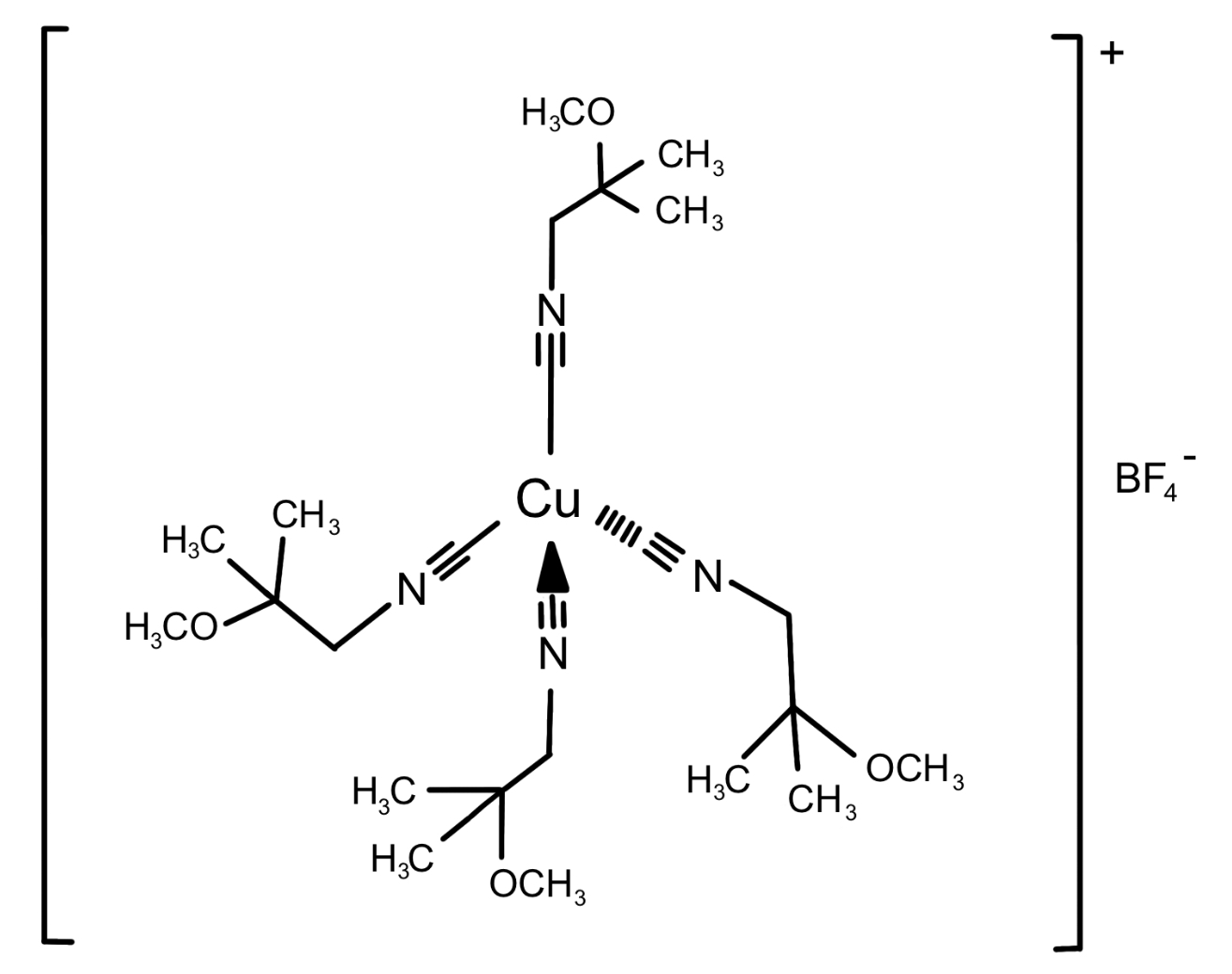
Kupfertetramibitetrafluoroborat (Summenformel: C24H44BCuF4N4O4) ist ein weißes bis fast weißes, kristallines Pulver mit einer molaren Masse von 603 g/mol. Es ist in Wasser schwer löslich und in wasserfreiem Ethanol leicht löslich. Bei der betrachteten Substanz handelt es sich um einen Komplex, welcher Kupfer als Zentralatom enthält und vier Methoxyisobutylisonitril-Liganden (MIBI) besitzt. Als Anion liegt Tetrafluoroborat vor, welches zu einer erhöhten Löslichkeit führt. Gleichzeitig stellt dieses Anion keinen Störfaktor für mögliche Reaktionen dar, da es in Reaktionen weitgehend durch seine niedrige Nucleophilie inert ist. Der Komplex ist stabil, besitzt keinen Geruch und hat keine toxischen Eigenschaften. Die Schmelztemperatur beträgt 96 °C bis 102 °C und der Gehalt der wasserfreien Substanz sollte entsprechend den Vorgaben der Arzneibuchmonographie bei 10,04 % bis 11,04 % Cu liegen. 2
(T-4)-Tetrakis[1-(isocyan-κC)-2-methoxy-2-methylpropan]kupfer(I)-tetrafluoroborat wird in der radiologischen Diagnostik verwendet, zum Beispiel zur Herstellung von Injektionslösungen, wie die ebenfalls im Europäischen Arzneibuch erwähnte [99mTc] Technetium-Sestamibi-Injektionslösung. 3 Dabei ist Kupfertetramibitetrafluoroborat eine Komponente eines Kits, welches zusätzlich Natriumcitrat, Mannitol, Zinn(II)chlorid-Dihydrat und L-Cysteinhydrochlorid-Monohydrat enthält. Dieses Kit wird im Zuge der radiologischen Diagnostik verwendet, um 99mTc-Komplexe im Rahmen einer reduktiven Synthese zu gewinnen. 4 Die resultierenden 99mTc-Komplexe werden anschließend als Tracer für bildgebende Verfahren verwendet, da sie kurzwelliges Licht in Form von Gammastrahlung emittieren. 5 Mögliche Untersuchungsmethoden sind zum Beispiel die Herzuntersuchung (Myokard-Perfusions-Szintigraphie) oder die Untersuchung von Nebenschilddrüsengewebe. 6
Durchführung der Monographie
Zur Gehaltsbestimmung des Schwermetallkomplexes Kupfertetramibitetarfluoroborat wird eine Massenspektrometrie mit induktiv gekoppeltem Plasma (ICP-MS) durchgeführt. Diese Art der Massenspektrometrie zeichnet sich vor allem durch ihre hohe Robustheit und ihre Empfindlichkeit aus, weshalb sie zur Identifizierung und Quantifizierung von Schwermetallen eingesetzt wird und deshalb auch bei diesem Komplex Anwendung findet. Die Kalibrierung erfolgt durch die Interne-Standard-Methode, weil es in Folge von Interferenzen zu Signal-Überlagerungen kommen kann.
Scandium wird zum Beispiel als Interner Standard eingesetzt, da durch die zum Teil geringe Nachweisgrenze keine Spuren davon in der Probe enthalten sein dürfen. Für die Herstellung der Prüflösung einer Konzentration von 0,8 mg ⋅ ml-1 erfolgt die Umsetzung der Prüfsubstanz mit Salpetersäure und Wasserstoffperoxid unter Einfluss von Wärme (Mikrowellenstrahlung). Die Konzentration der Kupfer-Stammlösung beträgt 1 ppm, welche aus einer Kupfer-Standard-Lösung (0,1 %), Salpetersäure und Wasser besteht. Zur Herstellung der Referenzlösungen dient die Stammlösung, welche eine Kupferkonzentration von +/- 20 % der in der Prüflösung zu erwartenden Kupferkonzentration enthalten soll. Die für die ICP benötigte Scandium-Lösung der Konzentration 10 ppm wird aus einer Scandium-Standardlösung der Konzentration 0,1 % hergestellt. 7
Auswertung/ Interpretation/ Bedeutung und Eignung der analytischen Methode
Durch die Zugabe von Scandium als internen Standard mit bekannter Masse lässt sich eine Aussage über den Probengehalt treffen. Das erhaltene Massenspektrum zeigt neben dem Molpeak des Analyten auch den des internen Standards. Wichtig bei der Wahl des internen Standards ist, dass sich die Isotopenmuster des internen Standards und des Analyten nicht überschneiden. Es ist möglich die Intensitäten der beiden Molpeaks in ein Verhältnis zu setzen und den Gehalt des Analyten zu bestimmen, da die Intensität des Peaks vom internen Standard proportional zu dessen Masse ist.
Die Quantitative Massenspektrometrie stellt eine einfache und sehr genaue Methode dar. Zudem ist sie für die Spurenanalytik geeignet, was bedeutet, dass auch Kleinstmengen analysiert werden können und somit keine großen Rohstoffmengen verbraucht werden. Die quantitative Bestimmung von Stoffen ist nur im linearen Bereich möglich. Vorteilhaft ist außerdem die Möglichkeit der Kopplung mit anderen Methoden, wie zum Beispiel GC-MS oder die hier erwähnte ICP-MS, durch welche eine hohe Selektivität und Empfindlichkeit erreicht werden kann. Dafür muss das Fragmentierungsmuster des Analyten bekannt sein. Ein Nachteil ergibt sich jedoch aus dem Mangel an Reproduzierbarkeit der Probenzuführung, welche es nötig macht mit einem internen Standard als Kalibriermethode zu arbeiten. 8
Einzelnachweise
1 Eigenes Design: L.Ungar ⇑
2 Kommentar zur Ph.Eur.9.2 Monographie: Kupfertetramibitetrafluoroborat zur Herstellung von radioaktiven Arzneimitteln 2547 ⇑
3 Ph.Eur. 10. Ausgabe, Grundwerk 2020 Monographie: Kupfertetramibitetrafluoroborat zur Herstellung von radioaktiven Arzneimitteln 2547 ⇑
4 Kommentar zur Ph.Eur.9.2 Monographie: Kupfertetramibitetrafluoroborat zur Herstellung von radioaktiven Arzneimitteln 2547 ⇑
5 https://de.wikipedia.org/wiki/Technetium (Abgerufen 06.05.2021; 09:42 Uhr) ⇑
6 https://studylibde.com/doc/8933255/produkt%C3%BCbersicht---rotop-pharmaka-gmbh (Abgerufen 05.05.2021; 19:44 Uhr) ⇑
7 Kommentar zur Ph.Eur.9.2 Monographie: Kupfertetramibitetrafluoroborat zur Herstellung von radioaktiven Arzneimitteln 2547 ⇑
8 Dominik, A., & Steinhilber, D., & Wurglics, M. (2013). Instrumentelle Analytik kompakt (3. Aufl.). S. 191. Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft. ⇑
TU-Braunschweig Institut für Medizinische und Pharmazeutische Chemie Seminar: Instrumentelle Analytik Kontakt: tubs@t-kellner.de
