HPLC
Expertenbericht zum Praktikumsversuch
HPLC
WiSe 2021/2022
Abgabedatum
08.12.2021
Expertengruppe 13
Ronja Müller
Linda Rottmann
Nicole Ernst
Identifizierung und Quantifizierung von Arzneistoffen (HPLC)
Inhaltsverzeichnis

Einleitung
Die Hochleistungs-Flüssigkeitschromatographie (englisch: High performance liquid chromatography, HPLC) ordnet sich den chromatographischen Trennmethoden zu und findet ihren Einsatz unter anderem in der Arzneimittelkontrolle, in der sie für die Bestimmung von Identität, Reinheit und Gehalt verwendet wird. So kann durch Trennung von Substanzgemischen eine qualitative, sowie eine quantitative Aussage darüber getroffen werden, welche Substanzen und zu welchem Anteil diese in der vorliegenden Probe vorhanden sind. In diesem Versuch kommt als Trennmechanismus die Umkehrphasenchromatographie zum Tragen, bei welcher unpolare Substanzmoleküle mit der unpolaren Oberfläche der stationären Phase wechselwirken. 2
Versuchsbeschreibung
Ziel des Praktikumsversuchs:
Mithilfe des HPLC-Systems (siehe Abb.1) wird in diesem Versuch ein Arzneistoffgemisch in seine Einzelkomponenten getrennt. Die Ermittlung der Identitäten erfolgt in der Auswertung über Vergleich der Retentionszeiten mithilfe von Referenzsubstanzen (siehe 1.5 Durchführung). Die Probe ist hinsichtlich der Zusammensetzung zu untersuchen und kann Acetylsalicylsäure (ASS), Paracetamol (PCM) und Coffein enthalten. Mit Hilfe von Kalibriergeraden wird der Gehalt der einzelnenen Substanzen bestimmt. Ebenfalls werden Trennfaktor und Retentionsfaktor der Peaks untersucht.3
Instrumenteller Aufbau

Abb.2 Aufbau der HPLC-Säulenchromatograhie 4 -- Die Urheberschaft an dieser Abbildung ist ungeklärt --

Als mobile Phase wird ein Gemisch aus Acetonitril und Wasser im Verhältnis 17,5% : 82,5% (V/V), mit einem pH-Wert von 2,5 eingesetzt. Um einen pulsationsfreien, kontinuierlichen Eluentenfluss zu gewährleisten, wird eine Doppelkolbenpumpe eingesetzt (siehe Abb.3). Die im Autosampler befindlichen Proben werden, mit Hilfe einer HPLC-Spritze (siehe Abb.4), in die Dosierschleife eingebracht. Durch das automatische Umschalten des Sechswegeventils in die Injektionsstellung gelangt die Probe in den Eluentenstrom. Das Injektionsvolumen beträgt hier 20 µl. Das Elutionsmittel wird zusammen mit der Probe, bei einer Flussrate von 1,25 ml/min, zur Säule transportiert. Als stationäre Phase wird eine Umkehrphase (RP-18) mit einer Partikelgröße von 5 µm verwendet. RP-18 steht für reversed-phase, dabei ist das Silicagel mit C18-Ketten modifiziert. Dadurch ist die Säulenoberfläche unpolar und dient zur Trennung von unpolaren Substanzen. Die Trennsäule (siehe Abb.5) ist 125 mm lang und der Innendurchmesser beträgt 4 mm. Nach der chromatographischen Trennung gelangen die Substanzen zum Detektor und werden durch einen UV-Detektor bei 207 nm detektiert. Das elektrische Signal wird anschließend von einem Computer (Interface, siehe Abb. 1) erfasst. Nach 12 Minuten wird die Messung automatisch beendet. 6 7
Die genaueren Beschreibungen der Einzelkomponenten sind im Artikel zur Hochleistungsflüsskeitschromatograhie zu finden. 8


Durchführung
Die Inbetriebnahme des HPLC- Messgerätes, sowie dessen Einführung erfolgt vor Beginn des Versuchs durch einen Assistenten / eine Assistentin. Erst dann sind alle folgenden Schritte mithilfe der ausliegenden Arbeitsanweisung selbst durchzuführen. Zu Beginn sind alle Glasgeräte auf Unversehrtheit und mögliche Verunreinigungen hin zu überprüfen. So wird gewährleistet, dass Verdünnungen ordnungsgemäß und möglichst genau hergestellt und die verwendeten Lösungen nicht verunreinigt werden.
Herstellung der mobilen Phase
Zur Herstellung der mobilen Phase werden 175,0 mL Acetonitril in einem Messzylinder abgemessen und in ein 1 L Becherglas vorgelegt. Mit Wasser in HPLC- Qualität wird das Acetonitril zu circa 900 mL verdünnt. In einem separaten Becherglas wird Phosphorsäure 85% im Verhältnis 1:4 verdünnt. Hierzu reicht i.d.R. ein Ansatz der Lösung von 20 mL aus. Das Acetonitril/ Wasser- Gemisch soll nun auf einen pH-Wert von 2,5 eingestellt werden. Dazu werden mithilfe eines pH- Meters, welches zuvor nach ausliegender Arbeitsanweisung kalibriert wurde, nach und nach kleine Mengen der verdünnten Phosphorsäure zum Gemisch gegeben bis der gewünschte pH-Wert eingestellt ist. Danach wird die Lösung in einen 1 L- Messkolben überführt und mit Wasser (HPLC- Qualität) zur Eichmarke aufgefüllt. Anschließend wird die Lösung auf dem Ultraschallbad entgast.
Vermessung der unverdünnten Probe
Die ausgegebene Analysenlösung befindet sich in einem 100 mL Messkolben und wird mit der mobilen Phase zu 100,0 mL auf die Eichmarke aufgefüllt.


Mit der ersten unverdünnten Probelösung wird nun die erste Messung durchgeführt. Dazu wird ein Glasfläschchen (Vial, siehe Abb.6) zu dreiviertel der Gefäßhöhe mit der zu untersuchenden Lösung befüllt und anschließend mit einem Schraubdeckel verschlossen. In den Schraubdeckel wird eine Membran eingelegt, durch die die Spritze Lösung aus dem Gefäß entnehmen kann. Die blaue Seite der Membran zeigt hierbei zur Lösung. Das befüllte Vial ist dann in die dafür vorgesehene Position im Rack des Autosamplers (hinterste Reihe, ganz links, siehe Abb.7) einzusetzen. Die Messung kann daraufhin begonnen werden. Zunächst wird die Basislinie aufgenommen, da auch das Lösungsmittel eine Eigenabsorption aufweisen kann. Danach wird die Probe vermessen. Eine Messung dauert dabei 12 Minuten, wobei der Verlauf der Messung beobachtet werden soll und die Maxima der Peakhöhen abgelesen werden. Dazu bietet es sich an, an den Bereich, in dem sich die Maxima befinden, heranzuzoomen. Dazu mit der Linkstaste der Maus einen Kasten ziehen, der das Maximum des zu beurteilenden Peaks und die y- Achse auf derselben Höhe einschließt. Wurde dieser Bereich vergrößert, so kann man durch Anklicken des Peakmaximums eine Linie zur y- Achse ziehen und den Wert ablesen. Durch Rechtsklick wird dann wieder das gesamte Spektrum angezeigt.
Vermessung der Kalibrierlösungen
Die nächsten Messungen erfolgen nun mit drei aus der Kalibrierlösung hergestellten Verdünnungen. Dazu werden etwa 100 mg ASS, 50 mg PCM und 20 mg Coffein genau auf einer Analysenwaage mithilfe eines Wägeschiffchens eingewogen, in einen 100 mL- Messkolben überführt und nach Zugabe von circa 80 mL mobiler Phase im Ultraschallbad gelöst. Dabei ist darauf zu achten, dass der Kolben ohne Stopfen in das Ultraschallbad gestellt wird. Die Einwaage und Rückwaage werden notiert. Nach vollständiger Lösung der Referenzsubstanzen wird der Messkolben bis zur Eichmarke mit mobiler Phase aufgefüllt. Anschießend wird die Lösung nochmals geschüttelt, um Homogenität zu gewährleisten. Aus dieser werden nun drei Verdünnungen hergestellt. Dazu wird mithilfe einer Kolbenhubpipette jeweils 1,0 mL in einen 10 mL, in einen 25 mL und in einen 50 mL- Messkolben pipettiert und mit mobiler Phase auf die jeweiligen Eichmarken aufgefüllt. Anschließend werden diese im Ultraschallbad entgast. Jede der Lösungen wird nun in ein eigenes Vial gefüllt, mit einem Schraubdeckel mit eingelegter Membran verschlossen und diese in die vorgesehenen Positionen des Racks im Autosampler gestellt (siehe Abb. 7). Gemäß der Arbeitsanweisung werden diese nun nacheinander vermessen. Die Höhen der Substanzpeaks werden notiert.
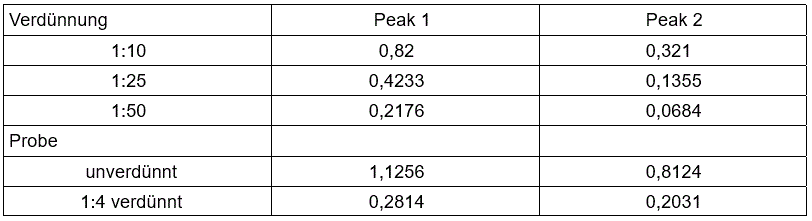
Vermessung der verdünnten Probe
Anhand der jeweiligen Höhen der Kalibrierlösungen ist die Verdünnung der Probelösung nun so zu wählen, dass diese eine Konzentration erhält, welche zwischen den Konzentrationen der Referenzlösungen liegt. Wurde diese rechnerisch ermittelt, so kann die Verdünnung hergestellt werden. Folgend ein Beispiel zur Veranschaulichung mit imaginären Werten (siehe Tabelle 1). Wird hier eine 1:4 Verdünnung gewählt, so fällt die Konzentration der Probelösung in den Konzentrationsbereich der Referenzlösungen. Die Probe wird daraufhin vermessen und die resultierenden Werte notiert.
Die Lösungen werden zum Schluss alle in dem Behältnis für organische Lösemittel entsorgt.14
Auswertung
Nach der Messung und vor Beginn der eigentlichen Auswertung wird für die Chromatogramme eine Korrektur der Basislinie vorgenommen, sofern dies nötig ist. Anschließend erhält man fünf verschiedene Auswertungsbögen (Probe unverdünnt; Referenzsubstanzen 1:10, 1:25, 1:50; Probe individuell verdünnt). In Abbildung 8 ist das Chromatogramm der 1:25 Verdünnung zu sehen. Relevante Messergebnisse sind die Totzeit (tM) und die Bruttoretentionszeiten (tR) der Substanzen, woraus die Nettoretentionszeit (tS) berechnet wird, sowie die dazugehörigen Areas.
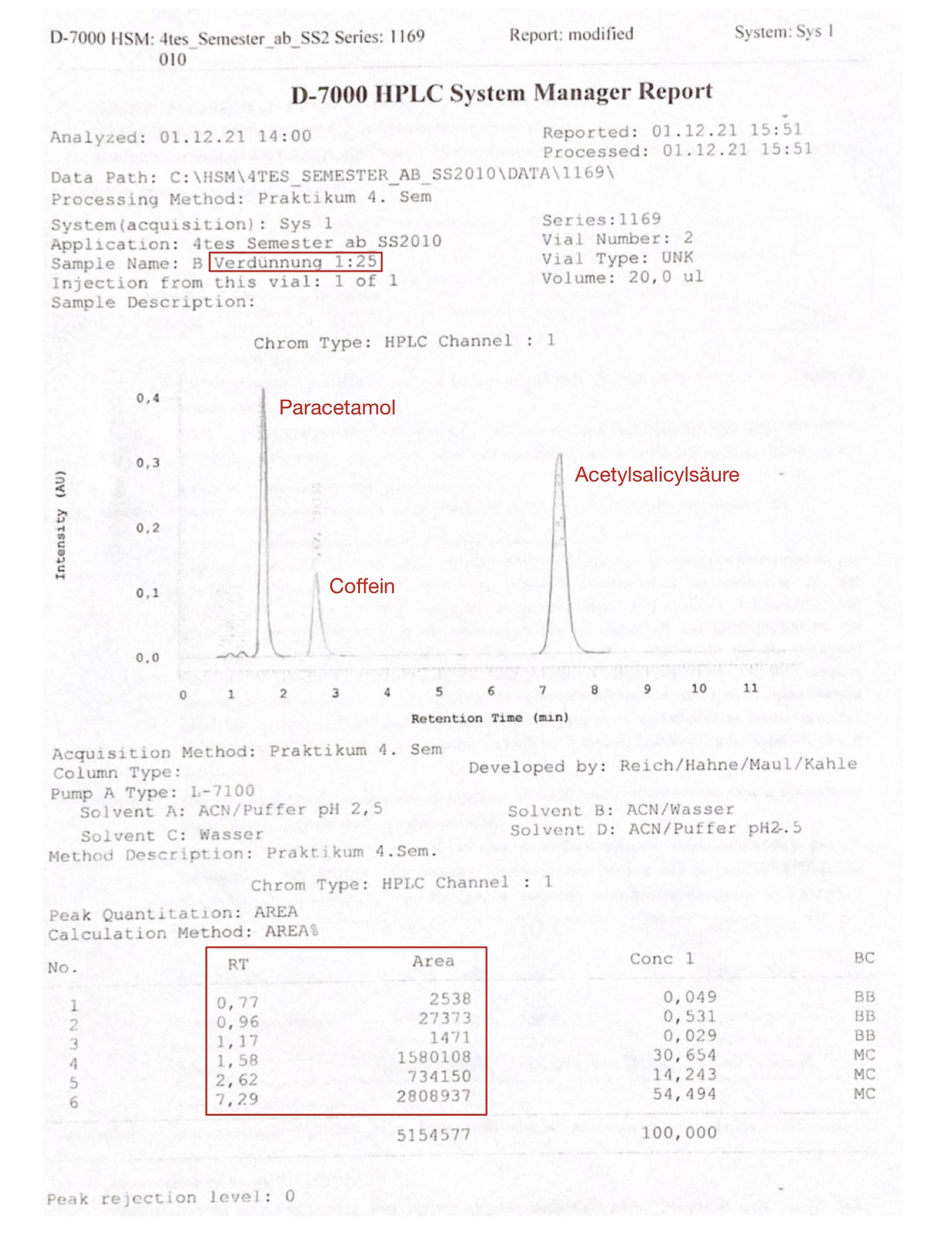
Die Identifizierung der enthaltenen Substanzen erfolgt durch Vergleich der Retentionszeiten mit Referenzsubstanzen.
Zur Quantifizierung werden die Areas benötigt. Die Berechnung der Konzentration in der Analysenlösung erfolgt durch Erstellung von Kalibriergeraden. Dabei wird für jede zu analysierende Substanz eine Gerade erstellt. Dazu werden auf der x-Achse die Massenkonzentrationen der Referenzlösungen (µg/ml) aufgetragen. Auf der y-Achse wird die dazugehörige gemessene Area aufgetragen. Die gemessene Fläche der verdünnten Probe wird in die erhaltene Geradengleichung eingesetzt, wodurch die unbekannte Konzentration des einzelnen Analyten bestimmt wird. Die Konzentration der Analyten wird jeweils in mg/100ml Analysenlösung angegeben.
Um die Messdaten und somit das Trennergebnis beurteilen zu können, werden für den Versuch verschiedene chromatographische Parameter herangezogen. Der Trennfaktor α wird ermittelt, um eine Aussage darüber treffen zu können, wie gut zwei Analyten voneinander getrennt wurden. Dieser gibt das Verhältnis zwischen zwei Retentionsfaktoren k wieder. Der Retentionsfaktor k wiederum gibt das Verhältnis zwischen dem Aufenthalt in stationärer und mobiler Phase wieder. Mit Hilfe der gemessenen Totzeit und Bruttoretentionszeit wird die Nettoretentionszeit berechnet. Anschließend kann mit dem Ergebnis der Retentionsfaktor berechnet werden. Dies wird für jeden gemessenen Peak der Substanzen durchgeführt und man erhält mehrere Retentionsfaktoren, die man ins Verhältnis setzen kann, wodurch man schließlich den Trennfaktor berechnen kann. 16
- Die verwendeten Formeln zur Berechnung:
⚠ $$ t_S = t_R - t_M ⚠ $$
⚠ $$ k = \frac {t_S}{t_M} ⚠ $$
⚠ $$ α = \frac {k_2}{k_1} ⚠ $$
- tS= Nettoretentionszeit, tR= Bruttoretentionszeit, tM= Totzeitα= Trennfaktor, k= Retentionsfaktor
Weiterführende Informationen zu den Parametern sind dem Artikel zur Hochleistungsflüsskeitschromatograhie | Parameter zu entnehmen. 17
Zur Ansage des Versuchs werden die Identität sowie die Massenkonzentration [mg/100mL] der zwei Arzneistoffe in der Analysenlösung angegeben, zusätzlich die jeweiligen Nettoretentionszeiten und Retentionsfaktoren. Außerdem werden die ermittelten Trennfaktoren der Referenzsubstanzen angesagt. Alle Werte sind auf drei signifikante Stellen anzugeben.
Allgemeine Tipps
- beim Entgasen auf dem Ultraschallbad den Stopfen entfernen
- auf eine korrekte Verdünnung der Probe mit mobiler Phase (nicht mit Wasser) achten
- um die Peakhöhen während der Messung ablesen zu können, kann man heranzoomen und mit der Maus eine Linie zur Achse ziehen
- mischt man 1 Teil der Probe mit 3 Teilen der mobilen Phase, erhält man eine 1:4 Verdünnung (Beispiel: 25 mL Probe mit 75 mL mobiler Phase)
- bei der Korrektur der Basislinie heranzoomen, da man sonst nicht erkennt ob diese schräg verläuft oder gerade ist
- die Totzeit entspricht der Retentionszeit des 1. Peaks, diese sollte immer ähnlich sein (auf verschwundene Peaks und eventuelle Verunreinigungen achten)
Einzelnachweise
1 eigene Aufnahme; Urheber: Ronja Müller ⇑
2 Nadine Francke, Skript Instrumentelle Analytik Seminarteil High Performance Liquid Chromatography, S. 4ff ⇑
3 Dr. Thomas Kellner, Skript Instrumentelle Analytik WiSe- 2021/22 Version 09: (Corona), 22. Oktober 2021, S. 71 ⇑
4 Dominik, Steinhilber, Wurglics, Instrumentelle Analytik kompakt, 2013, 3. Auflage, S. 244 ⇑
5 eigene Aufnahme; Urheber: Ronja Müller ⇑
6 Dr. Thomas Kellner, Skript Instrumentelle Analytik WiSe- 2021/22 Version 09: (Corona), 22. Oktober 2021, S. 71 ⇑
7 Rücker, Neugebauer, Willems, Instrumentelle pharmazeutische Analytik, 2013, 5. Auflage, S. 474 ⇑
9 eigene Aufnahme; Urheber: Ronja Müller ⇑
10 eigene Aufnahme; Urheber: Ronja Müller ⇑
11 eigene Aufnahme; Urheber: Linda Rottmann ⇑
12 eigene Aufnahme; Urheber: Linda Rottmann ⇑
13 eigene Grafik; Urheber: Nicole Ernst ⇑
14 Dr. Thomas Kellner, Skript Instrumentelle Analytik WiSe- 2021/22 Version 09: (Corona), 22. Oktober 2021, S. 71 ⇑
15 eigene Aufnahme; Urheber: Ronja Müller ⇑
16 Nadine Francke, Skript Instrumentelle Analytik Seminarteil High Performance Liquid Chromatography, S. 9ff ⇑
