Faellung
Titelblatt
Expertenbericht zum Praktikumsversuch
Fällung
WiSe 2021/2022
Abgabedatum
16.12.2021
Expertengruppe 03
Brsis Yacoub
Carla Karam
Lina Gropengießer
Simultanbestimmung Chlorid/Iodid (Fällung)
Bestimmung des Gehaltes von Iodid und Chlorid durch Titration mit Silbernitratlösung bei potentiometrischer Indikation.
Inhaltsverzeichnis
Einleitung
Die Potentiometrie beruht auf der Messung der Potentialdifferenz zwischen einer Indikatorelektrode und einer Bezugselektrode im stromlosen Zustand. Das gemessene Potential hängt von der Konzentration der Elektrolytlösung ab. Es wird zwischen direktpotentiometrischen Bestimmungen und potentiometrischen Titrationen unterschieden. Bei einer potentiometrischen Direktbestimmung wird die Konzentration des Analyten direkt über das gemessene Potential einer ionenselektiven Elektrode bestimmt. Im Gegensatz dazu wird bei einer potentiometrischen Titration das Verfahren zur Indikation des Äquivalenzpunktes verwendet, da sich hier das Potential sprunghaft ändert. Auf diese Weise lassen sich unter anderem Säure-Base-, Redox-, komplexometrische Titrationen oder, wie in diesem Versuch, Fällungstitrationen durchführen.
Versuchsbeschreibung
In diesem Versuch soll der Gehalt von Kaliumiodid und Kaliumchlorid in der Probe simultan bestimmt werden. Dazu wird eine Fällungstitration mit Silbernitratmaßlösung durchgeführt. Die Indikation des Äquivalenzpunktes erfolgt potentiometrisch.
Theoretische Grundlagen
Die in der Lösung enthaltenden Iodid- und Chloridionen reagieren mit Silbernitrat aus der Maßlösung zu den schwerlöslichen Salzen Silberiodid und Silberchlorid. Diese fallen während der Titration aus, sobald das Löslichkeitsprodukt überschritten wurde. Die ablaufende Reaktion während der Titration lautet:
⚠ $$I^{-}+Ag^{+}\longrightarrow{AgI}\downarrow⚠ $$
⚠ $$Cl^{-}+Ag^{+}\longrightarrow{AgCl}\downarrow⚠ $$
Die Reihenfolge in der die Halogenide ausfallen wird durch die Löslichkeitsprodukte bestimmt. Zuerst fällt die schwerer lösliche Verbindung mit dem kleineren Löslichkeitsprodukt aus, da die Lösung früher gesättigt ist.
Silberiodid hat ein kleineres Löslichkeitsprodukt (⚠ $K_L=8\cdot10^{-17} mol^2/l^2, {25 °C}$) als Silberchlorid (⚠ $K_L=2\cdot10^{-10} mol^2/l^2, {25 °C})$ und fällt somit im Versuch als Erstes aus.2 Da sich die beiden Löslichkeitskeitsprodukte hinreichend unterscheiden, können die beiden Halogenide simultan bestimmt werden.
Das an der Indikatorelektrode gemessene Potential ist abhängig von der Aktivität der Silberionen in der Lösung. Der potentialbildende Vorgang lautet:
⚠ $$Ag\rightleftharpoons{Ag}^{+}+e^{-}⚠ $$
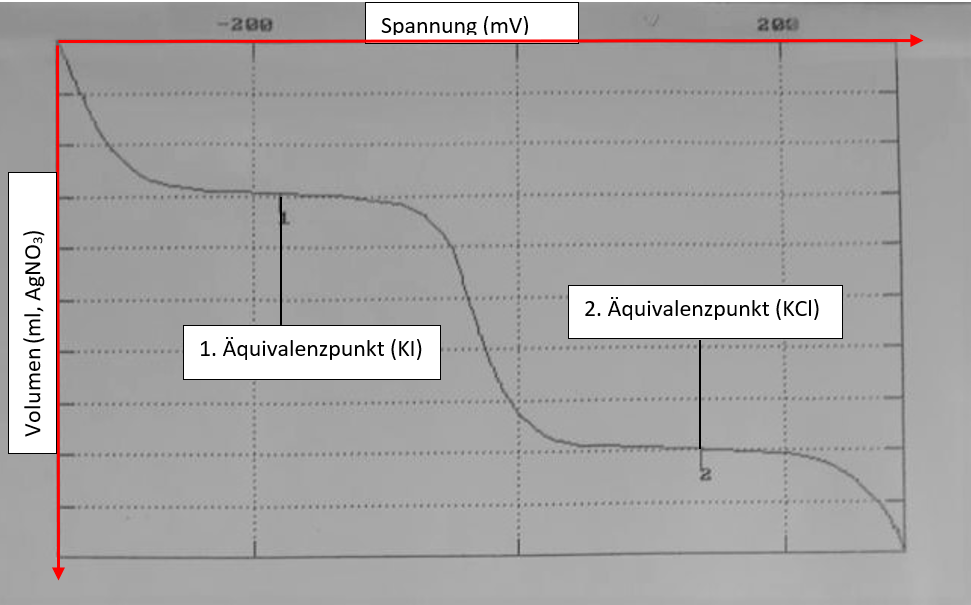
Nach dem Löslichkeitsprodukt ist nur eine begrenzte Menge von Silberiodid in der Lösung löslich. Zu Beginn enthält die Lösung noch eine hohe Konzentration an Iodidionen. Bei Zugabe von Silbernitrat reagiert der größte Teil der Silberionen mit Iodid und wird ausgefällt. Nur eine geringe Menge an Silberionen liegt gelöst vor. Am ersten Äquivalenzpunkt ist Iodid vollständig ausgefällt, die Konzentration der Silberionen in der Lösung nimmt schlagartig zu. Dies liegt daran, dass die Konzentration der gelösten Silberionen jetzt vom größeren Löslichkeitsprodukt von Silberchlorid abhängt. Durch die gestiegende Aktivität von Silberionen kommmt es zu einer sprunghaften Änderung des Potentials. Beim zweiten Äquivalenzpunkt ist auch das enthaltene Chlorid vollständig ausgefällt. Die Konzentration an Silberionen und damit auch das Potential nimmt wieder sprunghaft zu. In Abbildung 1 ist eine beispielhafte Titrationskurve vom Praktikumsversuch gezeigt, in der die Potentialsprünge an den Äquivalenzpunkten zu erkennenn sind. Bei der dargestellten Titrationskurve muss die korrekte Zuordnung der Achsen beachtet werden.
Instrumenteller Aufbau


Titrierautomat
In Abbildung 3 ist der Versuchsaufbau zu sehen. Durchgeführt wird die Titration mit einem Titrierautomaten. Dieser steuert automatisch die Zugabe an Maßlösung und ermittelt außerdem die Äquivalenzpunkte. Die Elektrode und die Bürette sind mit dem Titrierautomaten verbunden. Zur Titration werden diese in einer Halterung platziert, um in die Analysenlösung zu tauchen. Aus einem Vorratsgefäß wird die Maßlösung über die Bürette in die Analysenlösung geleitet. Zur Durchmischung der Analysenlösung ist ein Magnetrührer vorhanden. Auf dem Display wird während der Titration das gemessene Potential sowie das zugegebende Volumen der Maßlösung angezeigt. Die Äquivalenzpunkte lassen sich nach Ende der Titration hier anzeigen. Die Bedienung des Titrierautomaten erfolgt über das Keyboard und die Tasten unter dem Display nach der ausliegenden SOP.
Elektrode
Als Indikatorelektrode wird eine Silberelektrode verwendet und als Referenzelektrode eine Silber-Silberchlorid-Elektrode mit Kaliumnitrat als Zwischenelektrolyt. Beide sind zu einer Einstabsmesskette verbunden (siehe Abbildung 2). Die Silber-Silberchlorid-Elektrode enthält selbst eine Kaliumchloridlösung. Es muss verhindert werden, dass diese über das Diaphragma in die Probelösung austritt. Die Chloridionen würden sonst ebenfalls mit Silbernitrat reagieren und bei der Titration miterfasst werden. Deshalb enthält die Elektrode zusätzlich Kaliumnitrat als Zwischenelektrolyt, wodurch eine Vermischung verhindert wird.5
Das Phasendiagramm lautet:
⚠ $$ Ag|Ag^{+}|| KNO_3 ||Cl^{-},AgCl |Ag ⚠ $$
Durchführung


Es ist sinnvoll mit dem Lösen des Bariumnitrats zu beginnen, da dieses einige Zeit dauert. Dafür werden 1 g Bariumnitrat in ein Becherglas eingewogen. Dazu werden 0,5 ml Salpetersäure einer Konzentration von 2 mol/l mit einer Messpipette hinzugegeben und mit ungefähr 25 ml demineralisiertem Wasser gelöst. Der Lösevorgang wird mit einem Magnetrührer beschleunigt.
Die Zugabe von Salpetersäure verhindert, dass Silberhydroxid ausfällt, was bei einem pH-Wert im Alkalischen möglich wäre. Das hinzugegebene Bariumnitrat reagiert selektiv mit Chlorid-Ionen. So wird verhindert, dass Silberchlorid frühzeitig während der Fällung des Silberiodids mit ausfällt und es so zur Entstehung von Mischkristallen kommt. Dies würde zu fehlerhaften Messergebnissen bei der Bestimmung der Massenkonzentrationen führen. 8
Die Analysenlösung wird mit demineralisiertem Wasser bis zum Eichstrich des Messkolbens genau auf 200,0 ml aufgefüllt. Davon werden 25,0 ml mit einer Vollpipette entnommen und in das Becherglas mit der salpetersauren Bariumnitratlösung gegeben. Vor Beginn der Titration wird die Bürette gespült (über Taste DOS), damit Luftblasen entfernt werden. Die Bürette und die Elektrode werden in die Analysenlösung eingetaucht und sollten sich ungefähr auf gleicher Höhe befinden. Die Elektrode wird soweit eingetaucht, dass sich das Diaphragma der Elektrode vollständig in der Lösung befindet (siehe Abbildung 4). Das Ventil an der Elektrode ist während der Titration zu öffnen. In das Becherglas wird ein Rührfisch gegeben, sodass die Analysenlösung während der Titration mit dem Magnetrührer gerührt wird. Der Rührfisch soll dabei nicht gegen die Elektrode stoßen. Außerdem sollte die Rührgeschwindigkeit nicht zu niedrig eingestellt werden, damit sich der Niederschlag nicht stark an der Elektrode festsetzt. Anschließend kann die Titration mit Silbernitratmaßlösung (0,1 mol/l) über die Taste "Start" begonnen werden.
Der Titrierautomat titriert bis ein festgelegtes Volumen an Maßlösung hinzugegeben wurde. In der Zeit, in der der Automat eine Titration durchführt, kann bereits die Lösung für die nächste Titration vorbereitet werden. Abbildung 5 zeigt eine Probe nach abgeschlossener Titration mit dem ausgefällten Niederschlag.
Der Verbrauch an Maßlösung an den beiden Äquivalenzpunkten wird vom Titrierautomaten automatisch registriert. Da an dem Gerät kein Drucker angeschlossen ist, sollten die Werte nach einer beendeten Titration auf dem Display angezeigt und notiert werden. Die Anzeige der Werte erfolgt über das Keyboards nach der ausliegenden SOP. Insgesamt werden fünf Titrationen durchgeführt. Nach jeder Messung müssen Elektrode und Bürette durch Abspülen mit demineralisiertem Wasser und mit einem Papiertuch gesäubert werden, um Reste des Niederschlages zu entfernen.
Die austitrierten Lösungen werden im Abfallbehälter für wässrige Waschflüssigkeiten entsorgt. 9
Auswertung
Aus der Reaktionsgleichung geht hervor, dass die in der Maßlösung enthaltenen Silberionen mit den Iodid- und Chloridionen im Verhältnis 1:1 reagieren. An den Äquivalenzpunkten entspricht die Stoffmenge an zugegebenem Silbernitrat genau der enthaltenen Stoffmenge an Silberiodid oder Silberchlorid.
⚠ $${n(AgNO_3)= n(KI)}⚠ $$
⚠ $${n(AgNO_3)= n(KCl)}⚠ $$
Die Stoffmenge an Silbernitrat kann über die Konzentration, das hinzugefügte Volumen bis zum Äquivalenzpunkt sowie den Faktor der Maßlösung ermittelt werden:
⚠ $${n(AgNO_3)}= {c}\cdot{F}\cdot{V}⚠ $$
Durch Multiplikation mit der entsprechenden molaren Masse, erhält man die enthaltene Masse an Kaliumiodid oder Kaliumchlorid Es muss außerdem Verdünnung der Lösung berücksichtig werden. Da 25,0 ml der Lösung zu 100,0 ml verdünnt wurden,geschieht dies durch den Faktor 4.
Der Verbrauch an Maßlösung bis zum ersten Äquivalenzpunkt entspricht dem Gehalt an Kaliumiodid in der Probe. Die Formel zur Berechnung der Massenkonzentration von Kaliumiodid lautet somit:
⚠ $$\beta(KI) = 4\cdot{c}\cdot{F}\cdot{V(1.ÄP)}\cdot{M(KI)}⚠ $$
Die Differenz an Maßlösungsverbrauch zwischen dem ersten und dem zweiten Äquivalenzpunkt entspricht dem Gehalt an Chlorid. Die Berechnung für Kaliumchlorid erfolgt über die Formel:
⚠ $$\beta(KCl)=4\cdot{c}\cdot{F}\cdot(V(2.ÄP)-V(1.ÄP))\cdot{M(KCl)}⚠ $$
⚠ $c$: Konzentration Silbernitratlösung in mol/l
⚠ $F$: Faktor Silbernitratlösung
⚠ $V(1.ÄP)$: Verbrauch an Maßlösung bis zum ersten Äquivalenzpunkt in ml
⚠ $V(2.ÄP)$: Verbrauch an Maßlösung bis zum zweiten Äquivalenzpunkt in ml
⚠ $M$: Molare Masse in g/mol
⚠ $β$: Massenkonzentration in mg/100ml
Für jede der fünf Titration wird zunächst die Massenkonzentration berechnet und anschließend der Mittelwert daraus gebildet.
Außerdem erfolgt eine Berechnung der absoluten Standardabweichung und der relativen Standardabweichung. Die Standardabweichung ist ein Maß für die Streuung der Messwerte um den Mittelwert. Anhand der Standardabweichung lässt dich die Präzision einer Messung beurteilen. Eine Aussage über die Richtigkeit der Messung allein über die Standardabweichung kann nicht getroffen werden, da systematische Fehler nicht berücksichtig werden.10 Zum Vergleich von Standardabweichungen verschiedener Verfahren eignet sich die relative Standardabweichung. Da es sich um eine dimensionslose Größe handelt, können auch Daten verglichen werden, die in unterschiedlichen Einheiten gemessen wurden.11
Zur Berechnung der absoluten Standardabweichung wird die Formel für eine Stichprobe verwendet.
⚠ $$s=\sqrt\frac{\sum(x_i-\overline{x})^2}{n-1}⚠ $$
Die relative Standardabweichung berechnet sich aus dem Quotienten der absoluten Standardabweichung und dem Mittelwert. Die Angabe erfolgt in Prozent.
⚠ $${RSD\%}=\frac{s}{\overline{x}}\cdot100\%⚠ $$
⚠ $s$: absolute Standardabweichung der Massenkonzentration in mg/100 ml
⚠ $x_i$: einzelne Werte der ermittelten Massenkonzentrationen in mg/100 ml
⚠ $\overline{x}$: Mittelwert der Massenkonzentrationen in mg/100 ml
⚠ $n$: Anzahl der Titrationen
⚠ ${RSD\%}$: relative Standardabweichung der Massenkonzentration in Prozent
Einzelnachweise
1 eigene Aufnahme, Brsis Yacoub ⇑
2 https://www.formel-sammlung.de/formel-L%C3%B6slichkeitsprodukt-einiger-Salze-und-Hydroxide-4-37-207.html, zuletzt abgerufen am: 16.12.2021 ⇑
3 eigene Aufnahme, Brsis Yacoub ⇑
4 eigene Aufnahme, Lina Gropengießer ⇑
5 vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 568 ⇑
6 eigene Aufnahme, Brsis Yacoub ⇑
7 eigene Aufnahme, Lina Gropengießer ⇑
8 vgl. Orban,Dr. Oliver : Seminar: Instrumentelle Analytik, Einführung zu den praktischen Versuchen, Potentiometrische Fällung ⇑
9 vgl. Kellner, Dr. Thomas, Praktikumsskript Instrumentelle Analytik 4. Semester Pharmazie, Version:09 (Corona), Braunschweig 2021, S.27 ⇑
10 Wätzig, Prof. Dr. Hermann: Vorlesung Instrumentelle Analytik, Analytische Validierung, ICH-Guideline ⇑
11 https://www.medistat.de/glossar/deskriptive-statistik/variationskoeffizient, zuletzt abgerufen am 16.12.2021 ⇑

{kind=link}