Dünnschichtchromatographie
Bericht der Expertengruppe für
Dünnschichtchromatographie
SoSe 2021
Abgabedatum
21.06.2021
Über-/Expertengruppe 06
Maximilian Busse
Aaron Erfanian
Joel Fernandez
Judith Hillert
Franziska Reithmeier
Fiona Ringer
Dünnschichtchromatographie
Inhaltsverzeichnis
Einleitung
Die Dünnschichtchromatographie (DC) ist ein Verfahren, das die Trennung eines
Stoffgemisches durch unterschiedliche Wechselwirkungen mit einer stationären und einer
mobilen Phase ermöglicht. Sowohl qualitative als auch quantitative Bestimmungen können
durchgeführt werden.
Im Vergleich zu anderen chromatographischen Verfahren sticht die DC vor allem durch ihren einfachen apparativen Aufbau, der damit verbundenen Schnelligkeit, der Tatsache, dass zur Entwicklung einer DC-Platte nur sehr wenig Substanz benötigt wird und den geringen Kosten hervor. Alle Stoffgemische, die in Lösung gebracht werden können, können prinzipiell mithilfe der Dünnschichtchromatographie analysiert werden.
Grundlagen (physikalische/ chemische)
Das chromatographische System der DC besteht aus einer stationären und einer mobilen
Phase. Als stationäre Phase finden bestimmte Feststoffe Verwendung und als mobile Phase dienen flüchtige Flüssigkeiten.
Die Komponenten eines Substanzgemisches, das nun chromatographisch untersucht werden soll, wechselwirken aufgrund ihrer verschiedenen Eigenschaften unterschiedlich mit den beiden Phasen. In der DC spielen dabei v. a. Adsorptions- und Verteilungsprozesse eine große Rolle:
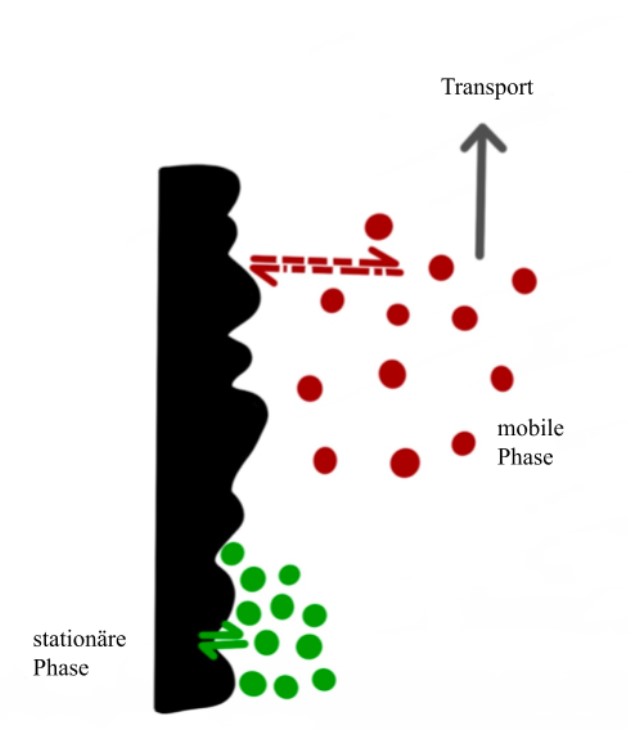
Adsorptionschromatographie
Bei der Adsorptionschromatographie kommt es zu Wechselwirkungen von Oberflächenstrukturen der stationären Phase mit funktionellen Gruppen des Analyten. Je nach Stärke der ausgebildeten reversiblen Bindung und je nach
Elutionskraft des Fließmittels (= mobile Phase), variiert die Geschwindigkeit, mit der ein Analyt
die DC-Platte (= stationäre Phase) passiert. Dieses Prinzip wird in der Abbildung 1 "Adsorptionsvorgänge" noch einmal verdeutlicht.
Komponenten, die schlecht mit der stationären Phase wechselwirken,
wandern zügig mit dem Fließmittel über die stationäre Phase hinweg und sind deshalb weit
oben auf der entwickelten DC-Platte zu finden. Komponenten, die gut mit der stationären Phase
wechselwirken, bleiben länger an ihr haften und liegen eher in der Nähe des Startpunktes.
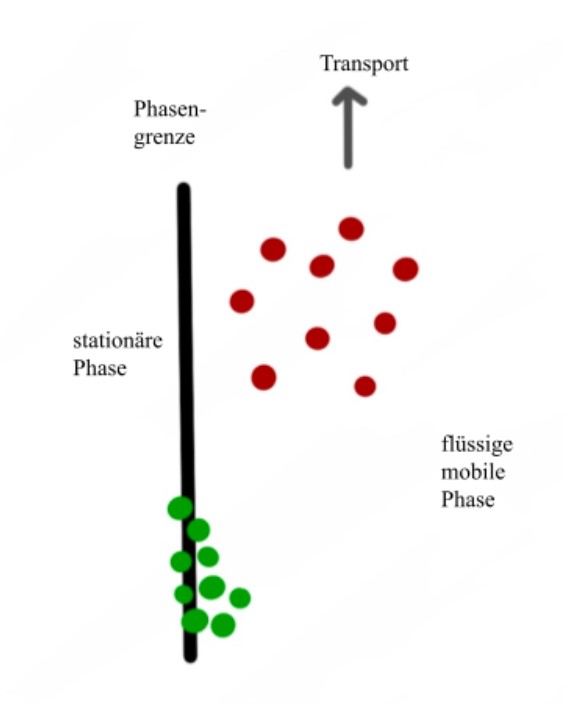
Verteilungschromatographie
In der Verteilungschromatographie ist es wichtig, dass stationäre und mobile Phase nicht mischbar sind.
Substanzen besitzen aufgrund ihrer chemischen Struktur verschiedene Lösungseigenschaften. Jede Komponente verteilt sich
deshalb unterschiedlich zwischen den beiden Phasen, sodass es zur Einstellung eines Verteilungsgleichgewichts kommt. Die Lage des Gleichgewichts hat auch hier zur Folge, dass die
Komponenten unterschiedlich schnell mit dem Fließmittel mitwandern und eine Trennung
der Komponenten erhalten wird. Ein Verteilungsgleichgewicht, das z. B. stark auf Seiten der mobilen
Phase liegt, sorgt dafür, dass die Komponente mit dem Fließmittel mitwandert und die DC-
Platte sehr zügig passiert, was die folgende Abbildung 2 "Verteilungsvorgänge" noch einmal zeigen soll. 2
Mithilfe des Nernst`schen Verteilungsgesetzes lässt sich die Verteilung zwischen den beiden Phasen beschreiben:
⚠ $$ K = \frac{c_{s}}{c_{m}}=\frac{V_{m} \cdot m_{s}}{V_{s} \cdot m_{m}} ⚠ $$
K: Verteilungskoeffizient
cs,m: Konzentration in der stationären/mobilen Phase
Vs,m: Volumen der stationären/mobilen Phase
ms,m: Masse der Komponente in der stationären/mobilen Phase
Stationäre Phasen
Stationäre Phasen in der DC sind i. d. R. feste Phasen. Meistens handelt es sich um eine dünne Schicht
aus Kieselgel, Kieselgur, Aluminiumoxid oder Cellulose 4, die auf einen Träger aufgebracht ist.
Um sich den Effekt der Verteilungschromatographie zu Nutze zu machen, kann die
stationäre Phase auch mit einem Flüssigkeitsfilm überzogen werden. Die verschiedenen
Platten unterscheiden sich zudem in Korngröße und Porenvolumen.
Kieselgel ist eine polare stationäre Phase, es handelt sich dabei um die sogenannte Normalphase. Die Adsorption an Kieselgel beruht auf der Wechselwirkung der polaren Silanolgruppen an der Oberfläche des Kieselgels mit polaren funktionellen Gruppen des Analyten.
Derivatisierte Kieselgele (RP = reversed phase, Umkehrphase) sind wesentlich unpolarer als
normales Kieselgel. Folglich wechselwirkt die stationäre Phase dann besser mit unpolaren
Analytmolekülen.
Es gibt noch viele weitere stationäre Phasen, siehe hierfür den Abschnitt "1.4 Instrumenteller Aufbau".
Es wird vermutet, dass eine stationäre Phase immer mit den Komponenten gesättigt
vorliegt, die am besten mit ihr wechselwirken. Das muss nicht unbedingt eine Komponente der
Probe sein, sondern kann auch eine Komponente des Fließmittelgemisches sein. Analyten
wechselwirken dann nicht direkt mit der stationären Phase, sondern mit ihrer „Belegung“. 5
Aufgrund dieser Wechselwirkung des Fließmittels mit dem Schichtmaterial der Platte (Sorbens) sollte weder das reine Fließmittel als mobile Phase, noch das reine Sorbens als stationäre
Phase bezeichnet werden.
Mobile Phasen
Eine wichtige Voraussetzung für die Durchführung einer DC ist, dass sich die zu untersuchenden Substanzen im Eluenten lösen.
Die mobile Phase zieht sich mithilfe von Kapillarkräften an der stationären Phase
entlang, trifft auf das aufgetragene Substanzgemisch und transportiert die Komponenten im
Fließmittelstrom weiter. Dabei herrscht ein ständiger Übergang der Analyten von der mobilen
in die stationäre Phase und umgekehrt, es tritt eine reversible Bindung der Analyten an die stationäre Phase auf.
Für diesen Effekt können Van-der-Waals-Kräfte, Dipol-Dipol-Wechselwirkungen, Wasserstoffbrückenbindungen, π-Komplexbildungen oder auch sterische Effekte verantwortlich sein. Diese Prozesse beeinflussen, wie bereits ausführlich beschrieben, den Auftrennungsprozess des Substanzgemisches.
Die verschiedenen Fließmittel können in eine eluotrope Reihe, das heißt in eine Ordnung von
Fließmitteln gemäß ihrer Polarität bzgl. einer bestimmten stationären Phase 6, eingeteilt
werden. Beispielsweise ist Methanol ein stärkeres Elutionsmittel als n-Hexan, wenn eine Normalphase verwendet wird.
Die Elutionskraft ist die Fähigkeit der mobilen Phase, einen Analyten von der
stationären Phase zu lösen.
Wechselwirkt z. B. eine polare Substanz mit der stationären Phase als Normalphase, dann
muss das Fließmittel so angepasst werden, dass es in der Lage ist, um die Bindestellen an
der stationären Phase zu konkurrieren und die Substanz von der stationären Phase zu lösen,
sodass die Substanz an der stationären Phase entlang transportiert werden kann.
Zu unpolare mobile Phasen könnten die Substanz in diesem Fall nicht über die DC-Platte
transportieren, sie würde am Startfleck „kleben“ bleiben. Allerdings darf das Fließmittel auch
nicht zu polar sein, da die Substanz sonst gar nicht mit der stationären Phase wechselwirken
kann und sofort „eluiert“ werden würde.
Fließmittel sind deshalb Lösemittel oder Lösemittelgemische verschiedener
Zusammensetzung und werden immer an das Trennproblem, die zu trennenden Komponenten
und die stationäre Phase angepasst, und evtl. auch nach einem vollständigen Durchlauf nochmal verändert, um
eine optimale Auftrennung zu erhalten.
Im Gegensatz zur Säulenchromatographie lässt sich hier die Fließgeschwindigkeit des
Fließmittels nicht so einfach anpassen. Das liegt daran, dass das Fließmittel in der DC nicht durch den Druck einer Pumpe bewegt wird, sondern sich alleine aufgrund der herrschenden Kapillarkräfte durch die stationäre Phase zieht. 7
Auswertung und Ergebnisse einer DC
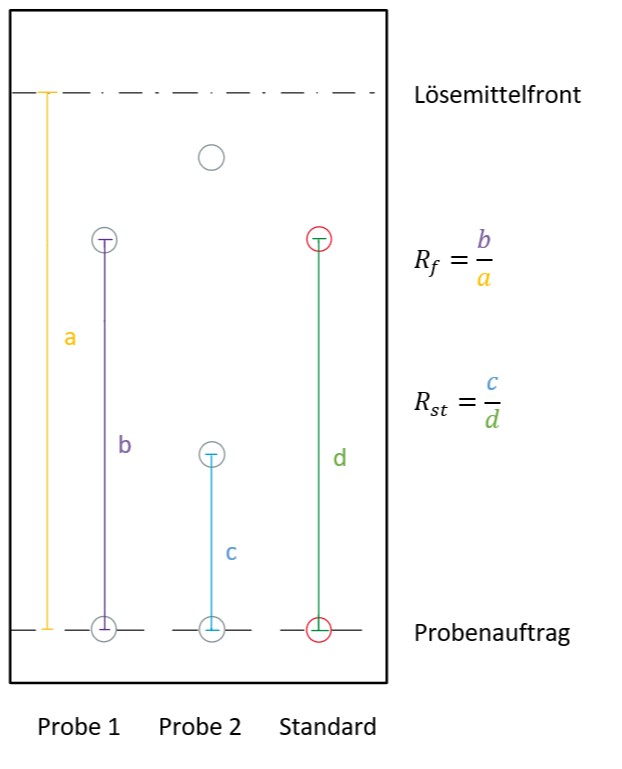
Als Resultat der DC wird der Rf-Wert (Retentionsfaktor, „retarding front“) angegeben.
Je nachdem, wie weit eine Komponente gewandert ist, zeigt sie einen Rf-Wert im Bereich
zwischen 0 und 1. Zur Berechnung dieses Werts wird die Entfernung der Komponente zum
Startpunkt mit der Entfernung der Laufmittelfront zum Startpunkt ins Verhältnis gesetzt. Da
eine Substanz nicht schneller wandern kann als das Fließmittel, kann der Rf-Wert maximal 1
betragen.
⚠ $$ R_{f} = \frac{Entfernung \: der \: Komponente \: zum \: Startpunkt}{Entfernung \: der \: Laufmittelfront \: zum \: Startpunkt}\ ⚠ $$
Je stärker eine Komponente von der stationären Phase "festgehalten" wird, desto weniger weit
wandert sie und desto geringer ist folglich ihr Rf-Wert.
Der Rf-Wert hängt von vielen Parametern (u. a. der stationären Phase, der Temperatur, des
Fließmittelgemisches, der Kammersättigung, etc.) ab und ist deshalb nur im selben
chromatographischen System eine Stoffkonstante.
Um unabhängigere/reproduzierbarere Aussagen treffen zu können, gibt es die Möglichkeit, eine Standardsubstanz mitlaufen zu lassen. Die Laufweite der Komponente wird dann nicht relativ zur Laufmittelfront, sondern zur Laufweite der Standardsubstanz angegeben. Dieser Wert wird als RSt-Wert bezeichnet9:
⚠ $$ R_{St} = \frac{Entfernung \: der\: Komponente\: zum \: Startpunkt}{Entfernung \: der \: Standardsubstanz \: zum \: Startpunkt}\ ⚠ $$
In der Abbildung 3 "Verbildlichung der Berechnung des Rf- und des RSt-Werts" ist diese Berechnung noch einmal veranschaulicht.
Instrumenteller Aufbau
Allgemeiner Aufbau
Für die Dünnschichtchromatographie wird der Analyt auf eine Platte aufgetragen, welche dann in die Chromatographiekammer (auch DC-Kammer oder Entwicklungskammer) mit dem Elutionsmittel gestellt wird.
Die Chromatographiekammer muss einen flachen Boden und einen dichtschließenden Deckel haben. Sie sollte aus einem durchsichtigem, inerten Material bestehen. Es ist wichtig, dass in der Kammer immer eine für die Kammergröße ausreichende und reproduzierbare Menge Fließmittel ist. Für die horizontale Entwicklung geeignete Kammern müssen außerdem über ein Behältnis für das Elutionsmittel mit einer Vorrichtung zur Auftragung des Elutionsmittels auf die stationäre Phase verfügen.10
Auf der Platte ist die stationäre Phase, ein Feststoff oder eine an Feststoff adsorbierte Flüssigkeit, aufgetragen. Die stationäre Phase (auch Sorbens) wird als dünne Schicht auf den Träger aufgebracht. Dies kann von Hand oder maschinell geschehen. Für schnellere Analysen und bessere Reproduzierbarkeit werden jedoch industriell gefertigte und standardisierte Fertigplatten verwendet. Verschiedene Platten unterscheiden sich durch den Plattenuntergrund, das Schichtmaterial, die Korngröße, die Schichtdicke und Zusätze in der Schicht. Die Träger bestehen aus Glas oder Kunststoff- bzw. Aluminiumfolien. Während Glasplatten zwar beständiger gegen hohe Temperaturen und aggressive Fließmittel sind, sind sie auch teurer als die Kunststoff- oder Aluminium-Alternativen. Folien hingegen haben außerdem den Vorteil, dass sie leicht in passende Formen zugeschnitten werden können. Gängige stationäre Phasen sind z. B. Kieselgel oder Aluminiumoxid. Das Kieselgel kann auch chemisch modifiziert werden, um eine noch bessere Trennung für bestimmte Analyten zu erzielen. Manche Platten müssen vor der Nutzung mit Lösemitteln gewaschen oder im Trockenschrank bei 120°C aktiviert werden. 11
Durchführung der DC
Das Auftragen der Analysensubstanz erfolgt mithilfe einer schmalen Kapillare. Hierbei sollte darauf geachtet werden, dass die Substanzflecken einen möglichst kleinen Durchmesser behalten, um eine deutliche Trennung zu erzielen. Außerdem müssen alle Substanzen auf der gleichen Höhe (Startlinie) und in möglichst regelmäßigen Abständen aufgetragen werden. Um dies zu erleichtern kann eine Auftragsschablone verwendet werden.12
Die Entwicklung kann entweder horizontal oder vertikal erfolgen.
Für die vertikale Entwicklung wird die Kammer zuerst mit Filterpapier ausgekleidet, dann das Elutionsmittel hinzugegeben und der Deckel der Kammer verschlossen.
Für eine vollständige Sättigung der Kammer wird diese eine Stunde bei Standardbedingungen (20°C) stehen gelassen. Sofern nicht anders vorgegeben, sollte die DC in einer gesättigten Kammer stattfinden. Anschließend wird die vorbereitete Platte möglichst senkrecht in die Kammer gestellt, wobei darauf geachtet werden sollte, dass das Fließmittel unter der Startlinie liegen sollte, an der die Proben aufgetragen sind. In der geschlossenen und vor Licht geschützten Kammer legt das Fließmittel die vorgeschriebene Laufstrecke zurück. Daraufhin wird die Platte entfernt und getrocknet sowie die Fließmittelfront markiert. Die Chromatogramme werden je nach Vorschrift über unterschiedliche Wege der Detektion sichtbar gemacht.
Für die horizontale Entwicklung wird eine Chromatographiekammer mit einer Wanne benötigt. In diese wird die Platte waagerecht hineingelegt. Das Elutionsmittel wird aus der Wanne auf die stationäre Phase geleitet, bis die nötige Laufstrecke zurückgelegt wurde. Dann kann die Platte herausgenommen und das Chromatogramm sichtbar gemacht werden. 14
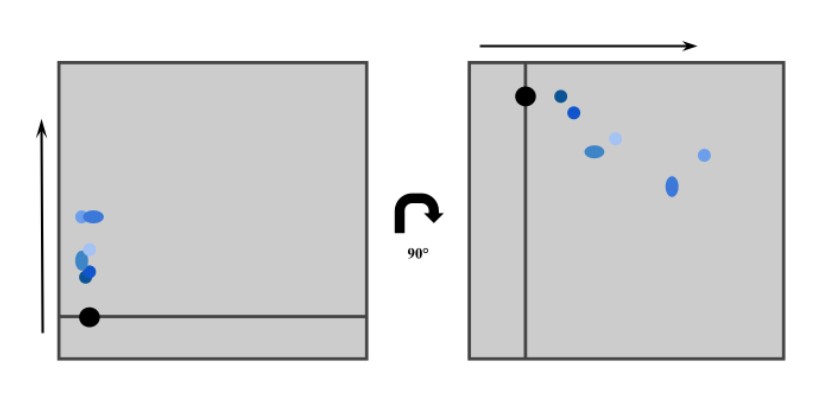
In der Regel wird in der Dünnschichtchromatographie mit einer eindimensionalen Trennung gearbeitet, bei der die Substanz einen Laufweg zurücklegt. Für bestimmte Substanzen und Anwendungen kann jedoch auch eine zweidimensionale Trennung nützlich und notwendig sein. Hierbei wird die Platte nach der ersten Entwicklung getrocknet, dann um 90° gedreht und erneut entwickelt. Bei der zweiten Trennung kann entweder das gleiche oder ein anderes Elutionsmittel gewählt werden. Dasselbe Elutionsmittel empfiehlt sich für Substanzen, die sich im Laufe der DC verändern, während unterschiedliche Elutionsmittel bei unveränderten Substanzen eine bessere und z. T. deutlichere Trennung erzielen können.
Detektion
Vor der Detektion muss die Platte vollständig von Fließmittel-Resten befreit werden, was i. d. R. mit einem Föhn unter dem Abzug geschieht.
Die Detektion durch Anregung zur Fluoreszenz kann z. B. bei Alkaloiden, Cumarinderivaten oder Salicylsäurederivaten unter UV-Licht (366 nm) genutzt werden. Die Substanzen heben sich als fluoreszierende Flecken auf dem dunklen Plattenuntergrund ab.15
Die Luminsezenzminderung als Detektionsmethode kann auf bestimmten Platten mit einem Fluoreszenz- oder Phosphoreszenzindikator genutzt werden. Hierbei wird ausgenutzt, dass z. B. aromatische Verbindungen UV-Strahlung bestimmter Wellenlänge (254 nm) absorbieren können, während der Indikator auf der Platte diese fluoreszieren lässt. So werden die Substanzflecken als dunkle Stellen auf der sonst leuchtenden Platte sichtbar.16
Bei der Nutzung von Detektionsreagenzien wird die Platte mit dem entsprechenden Reagenz besprüht. Dabei darf die Platte jedoch nicht zu feucht werden, um ein Verlaufen der Flecken zu verhindern. Durch Reaktion der Substanz(en) mit dem Detektionsreagenz entstehen gefärbte Flecken, welche dann ausgewertet werden können.17
Nachdem alle Substanzflecken sichtbar gemacht wurden, werden sie mit einem Bleistift markiert und ausgewertet. Zur Auswertung werden außerdem die Startlinie der Substanzen und die Laufmittelfront markiert. Ausgehend von den markierten Punkten können jetzt die der Retentionsfaktor Rf und der relative Retentionsfaktor RSt berechnet werden.
HPTLC
Die HPTLC (High Performance Thin-Layer Chromatography; dt. Hochleistungsdünnschichtchromatographie) ist eine Weiterentwicklung der DC. Sie hat eine deutlich höhere Trenneffizienz, Präzision und Detektierbarkeit. Ausschlaggebend für die Leistungsstärke der HPTLC sind vor allem die Trennschichten, welche durch kleinere Korngrößen, engere Korngrößenverteilung und eine homogenere Schichtdicke für bessere Trennungen sorgen. Aufgrund der hohen Trenneffizienz werden für die HPTLC auch nur Probenmengen im nanogramm-Bereich und relativ kurze Trennstrecken benötigt. Außerdem werden HPTLC-Platten meistens horizontal entwickelt.18
Quantitative DC
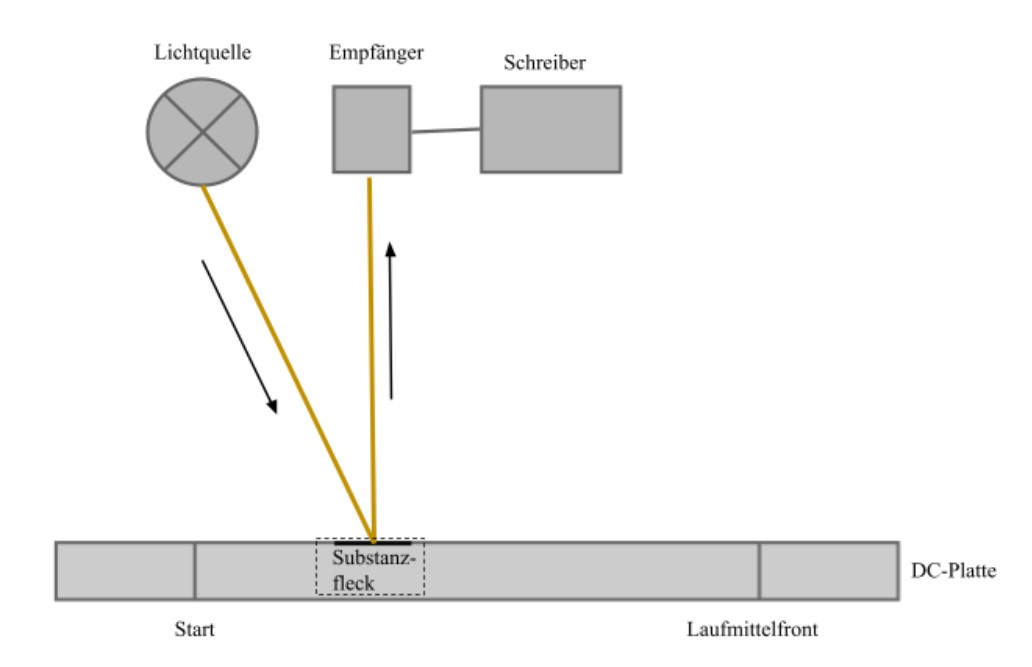
Bei der Durchführung einer quantitativen Analyse mittels DC werden sog. DC-Scanner genutzt, welche ein Photometer und eine Vorrichtung zum Bewegen der Platte durch den Lichtstrahl enthalten. Außerdem werden wegen ihrer erhöhten Trennleistung und ihrem geringerem Untergrundsignal HPTLC Platten verwendet.
Das monochromatische Licht aus der Lichtquelle des Photometers trifft unter einem bestimmten Winkel auf die Platte und wird von dort diffus reflektiert. Die Messung ist abhängig von der Remission bzw. Remissionsminderung des Analyten und Fließmittels. Remission bedeutet, dass ein Teil des Lichtspektrums absorbiert und ein Teil reflektiert wird. Sie wird von einem Empfänger gemessen. Während der Messung wird die DC-Platte langsam in Laufrichtung des Elutionsmittels bewegt. An den Stellen, an denen auf der Platte nur das Elutionsmittel vorliegt, wird ein Großteil des Lichtes reflektiert und es kommt nur zu einer geringen Remissionsminderung. Trifft das Licht auf einen Substanzfleck, wird ein Teil des Lichtes absorbiert, sodass es zu einer stärkeren Remissionsminderung kommt. Diese Minderung ist mengenabhängig und kann daher für die quantitative Analyse genutzt werden. 20
Mit Hilfe eines Computers wird eine Orts-Remissionskurve erstellt und ausgewertet. Zur Quantifizierung wird die Peakhöhe benutzt, da sie im Falle der Hochleistungsdünnschichtchromatographie genauere Werte liefert als die Peakfläche. 21 Die in DC-Scannern verwendete Remissionsminderung funktioniert nur, wenn die Analysensubstanz ein Chromophor besitzt, welches die Lichtenergie absorbiert, wodurch die Gesamtremission vermindert wird. Ein pharmazeutisch relevantes Beispiel ist das Malariamittel Chinin. 22
Da die Remissionsminderung wellenlängenabhängig ist, empfiehlt es sich für einzelne Substanzen zunächst passende Messwellenlängen zu ermitteln. Dafür wird ein Remissionsspektrum angelegt, indem ein Substanzfleck auf der HPTLC-Platte mit einer Vielzahl an Einzelwellenlängen beleuchtet und jeweils die Remissionsminderung gemessen wird. Die Wellenlänge, bei der sich ein Maximum ergibt, ist in der Regel eine geeignete Messwellenlänge. 23
Die Bestimmungsgrenzen liegen bei der quantitativen DC zum Teil im niedrigen Nanogramm Bereich24. Der hohe Probendurchsatz (bis zu 72 Proben auf einer HPTLC-Platte) erlaubt die Messung vieler Proben in kurzer Zeit. Das Verfahren ist zudem gut automatisierbar, was die quantitative Dünnschichtchromatographie zu einer leistungsstarken Methode zur Trennung und Quantifizierung von Substanzgemischen macht.25
Auswahl des Trennsystems
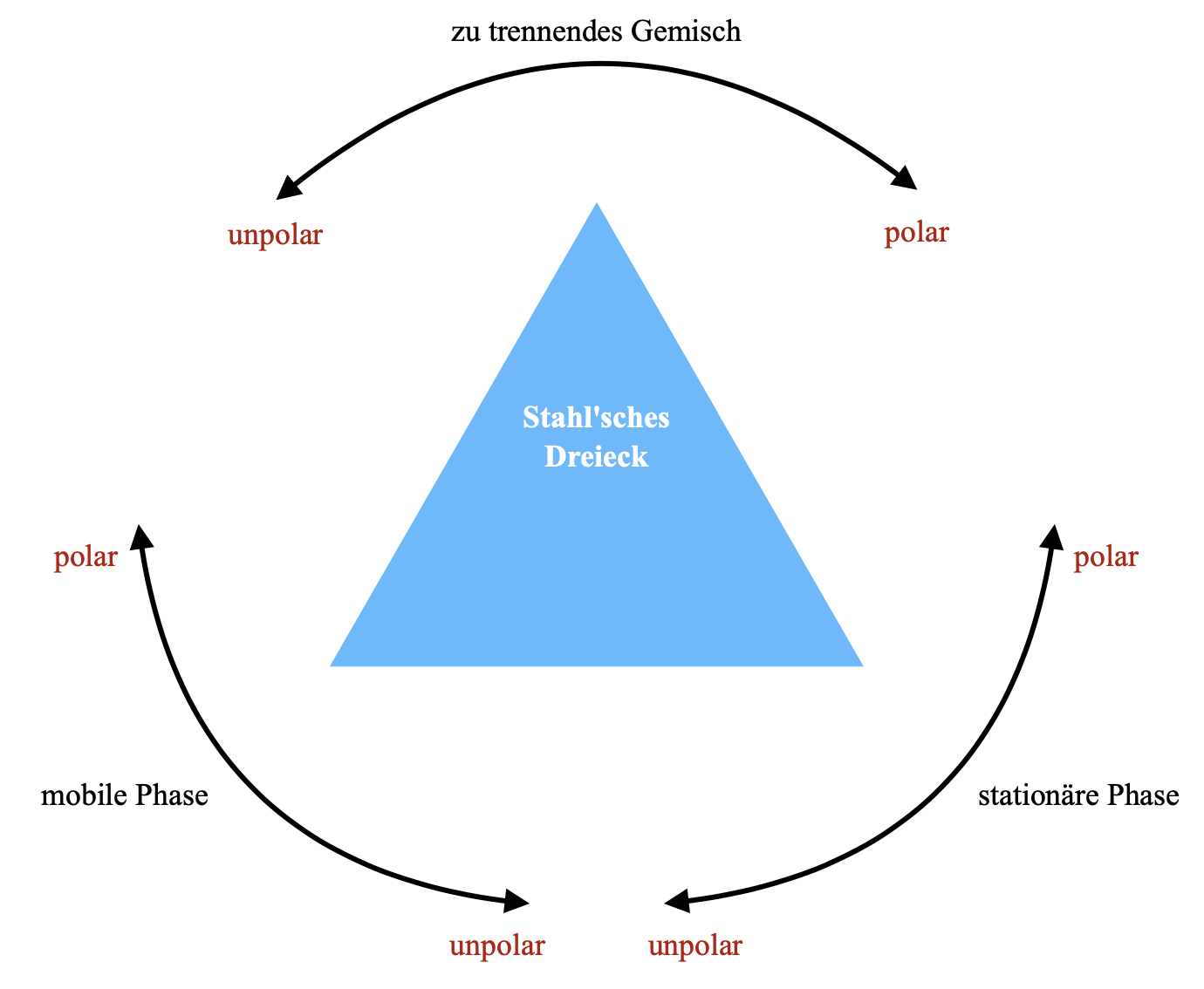
Die Auswahl von stationärer und mobiler Phase für die dünnschichtchromatographische Trennung ist immer abhängig vom Analysengemisch. Für Arzneistoffe sind Vorschriften in den Arzneibüchern sowie im Deutschen Arzneimittel-Codex zu finden.
Generell ist bei der Auswahl eines geeigneten Systems wichtig, dass die Substanzen ausreichend unterschiedlich reteniert werden, ohne zu nah beieinander zu liegen. Außerdem dürfen Substanzen nicht zu stark im Elutionsmittel eluieren, da sie noch einen ausreichenden Abstand zur Laufmittelfront behalten sollten, um vergleichbare Retentionsfaktoren zu liefern. Die Auswahl des Trennsystems kann mithilfe eines Stahl'schen Dreiecks erleichtert werden (s. Abbildung 6). 27
Des Weiteren muss bei sauren oder basischen Analysensubstanzen beachtet werden, dass es durch Dissoziation zu sog. Tailing (Schwanzbildung) kommen kann. Hierbei verschwimmen die Substanzflecken, da dissoziierte Teile des Analyten unterschiedlich im Fließmittel eluieren als nicht dissoziierte Teile. Dadurch wird die Trennung ungenauer und die einzelnen Flecken sind nicht mehr deutlich erkennbar. Um sicherzustellen, dass die Analysensubstanz vollständig undissoziiert vorliegt, sollten in geringen Mengen angemessene Modifier (Säuren oder Basen) dem Elutionsmittel zugesetzt werden. Bei sauren Substanzen wird häufig Ameisen- oder Essigsäure und bei basischen Substanzen Ammoniak oder Diethylamin als Modifier zugesetzt.28
Polare Komponenten im Fließmittel können zu ꞵ-Fronten führen, an denen sich Substanzen in Banden anreichern. An einer ꞵ-Front werden die polaren Komponenten des Fließmittels an die stationäre Phase adsorbiert und führen so zu einer heterogenen Fließmittelkomposition und somit ungenauen Trennung.29
Anwendung in der Pharmazie
Die Dünnschichtchromatographie findet vielfältige Anwendung in der pharmazeutischen Analytik. Sie wird sowohl für qualitative als auch quantitative Bestimmungen eingesetzt. Wichtige Anwendungsbeispiele sind:
- Identitätsprüfungen
- Trennungen von Enantiomeren
- Reinheitsprüfungen
- Drug Monitoring
- Qualitätskontrolle in der Arzneistoffproduktion
- Gehaltsbestimmungen von Arzneistoffen
- Untersuchung der Stabilität von Arzneistoffen
- Nachweis von Arznei- und Giftstoffen in Körperflüssigkeiten
Im Arzneibuch wird die Dünnschichtchromatographie aufgrund ihrer schnellen und unkomplizierten Durchführung sowie des geringen Probenverlustes oft zur Identitätsprüfung von organischen Arzneistoffen, Drogen und Drogenzubereitungen ergänzend zu anderen Methoden hinzugezogen.
Identitätsprüfungen können auf zweierlei Art erfolgen:
1. Vergleich der Probe mit einer Referenz auf derselben DC -Platte (!): Die Referenzlösung wird aus einer Substanz bekannter Identität hergestellt (sog. CRS = Chemische Referenz Substanz). Die Prüfung erfolgt visuell durch Vergleich des Auftrennungsmusters bzw. der Lage des jeweiligen Substanzflecks der Probe mit der Referenz
2. Relative Lage von dem Substanzfleck zu Vergleichssubstanzen (auf derselben DC-Platte): Diese wird durch den RSt - Wert (RSt = relativer Retentionsfaktor) beschrieben und bezieht sich, anders als der Rf - Wert, auf die relativ zum Referenzfleck (und nicht zur Fließmittelfront) zurückgelegte Strecke des Probenflecks. Zur Vereinfachung der Auswertung besitzen viele Drogen-Monographien im Arzneibuch schematische Abbildungen der Chromatogramme.
Chirale Trennungen lassen sich durchführen, indem die hydrophobierte Kieselgeslschicht (stationäre Phase) mit einem ebenfalls chiralen „Selektor“ imprägniert wird. Enantiomere Stoffgemische bilden mit diesem Selektor unterschiedlich starke, diastereomere Assoziate, wodurch sich die Enantiomere auftrennen lassen. Das Europäische Arzneibuch sieht die chirale Trennung für die Prüfung der Enantiomeren -Reinheit von L-([11C]Methyl)Methionin-Injektionslösung zu diagnostischen Zwecken vor.30
Halbquantitative Reinheitsprüfungen werden häufig wie folgt durchgeführt: Eine Probelösung mit der zu untersuchenden Substanz wird hergestellt. Außerdem wird eine Verdünnung der Probelösung, oft im Verhältnis 1:100 oder 1:200 hergestellt. Diese Verdünnung dient als Referenzlösung. Gleiche Volumina beider Lösungen (Probe und Referenz) werden auf die Platte aufgetragen und die Platte wird entwickelt.
Verglichen werden die Verunreinigung(en) in der unverdünnten Probelösung mit der Probensubstanz in der verdünnten Referenzlösung. Erstere dürfen keine stärkere Farbreaktion mit bestimmten Sprühreagenzien eingehen beziehungsweise darf der Fleck der Verunreinigung aus der Probenlösung (Untersuchung meist unter einer UV-Lampe) nicht größer sein als der der Probenfleck aus der Referenzlösung. Auf diese Weise wird der Gehalt von Verunreinigungen auf ca. 0,5- 1% begrenzt (Grenzwert variiert je nach Monographie).31
Die Methode ist zwar geeignet, um die Einhaltung bestimmter Grenzwerte abzuschätzen, anders als andere Methoden erlaubt sie jedoch keine genaue Quantifizierung der Verunreinigungen.
Drug Monitoring/ Qualitätskontrolle/Gehaltsbestimmungen:
Da die Dünnschichtchromatographie in Kombination mit geeigneten Detektoren (siehe oben) auch für quantitative Bestimmungen von sehr geringen Substanzmengen geeignet ist, kommt sie zum Beispiel für den Nachweis des Blutplasmaspiegels von Arzneistoffen zum Einsatz. Das entsprechende Verfahren heißt Drug Monitoring. Durch den hohen Probendurchsatz (siehe oben) sind auch Qualitätskontrollen im industriellen Maßstab mittels quantitativer HPTLC möglich. Ein Beispiel hierfür wäre die Gehaltsbestimmung von Tabletten.32
Für die Quantitative Dünnschichtchromatographie wird i.d.R die Remission einer Substanz erfasst und über diese die Stoffmenge ermittelt. Darüber hinaus beschreibt das Arzneibuch die Durchführung quantitativer Bestimmung auch für radioaktive Arzneistoffe
Das Arzneibuch sieht für quantitative Bestimmungen mittels Dünnschichtchromatographie eine Kalibrierung vor. Im Falle von Remissionsmessungen wird mit mindestens 3 Referenzen bekannter Stoffkonzentration, die 80%, 100% und 120% der erwarteten Stoffkonzentration der Probe entsprechen, kalibriert. Die Referenzen werden auf dieselbe Platte wie die Probe aufgetragen. Für Radiopharmaka wird eine Referenzprobe verwendet, die 100% der erwarteten Konzentration entsprechen soll. In diesem Fall wird keine Remission, sondern die radioaktive Strahlung gemessen.
Einzelnachweise
1 eigene Abbildung ⇑
2 Vgl. Dominik, Steinhilber, Wurglics (2013): Instrumentelle Analytik kompakt, S.200-203, S.206. 3. völlig neu bearbeitete und erweiterte Auflage. Wissenschaftliche Verlagsgesellschaft Stuttgart. ⇑
3 eigene Abbildung ⇑
4 Vgl. https://de.wikipedia.org/wiki/D%C3%BCnnschichtchromatographie (zul. gesehen am: 16.05.2021, 11:31) ⇑
5 Vgl. http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/croma/duennschichtchromatographie.vlu/Page/vsc/de/ch/3/anc/croma/dc/stat_phase/kieselgel/kisel1m70te0101.vscml.html (zul. gesehen am: 16.05.2021, 11:32) ⇑
6 Vgl. http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/croma/duennschichtchromatographie.vlu/Page/vsc/de/ch/3/anc/croma/dc/mob_phase/exp/laufmitteltest1m70te0101.vscml.html (zul. gesehen am: 16.05.2021, 11:32) ⇑
7 Vgl. http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/croma/duennschichtchromatographie.vlu/Page/vsc/de/ch/3/anc/croma/dc/prinzip/prinzip1m70te0101.vscml.html (zul. gesehen am: 16.05.2021, 11:32) ⇑
8 eigene Abbildung ⇑
9 Vgl. Dominik, Steinhilber, Wurglics (2013): Instrumentelle Analytik kompakt, S.238. 3. völlig neu bearbeitete und erweiterte Auflage. Wissenschaftliche Verlagsgesellschaft Stuttgart. ⇑
10 Vgl. Deutscher Apotheker-Verlag: Europäisches Arzneibuch, 10. Ausgabe, 1. Nachtrag: Amtliche deutsche Ausgabe (Ph. Eur. 10.1)., München, Deutschland: Deutscher Apotheker Verlag, 2021, 10.0/2.02.27.00. ⇑
11 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, 21.1.2. ⇑
12 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, 21.1.2. ⇑
13 eigene Abbildung ⇑
14 Vgl. Deutscher Apotheker-Verlag: Europäisches Arzneibuch, 10. Ausgabe, 1. Nachtrag: Amtliche deutsche Ausgabe (Ph. Eur. 10.1)., München, Deutschland: Deutscher Apotheker Verlag, 2021, 10.0/2.02.27.00. ⇑
15 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, 21.1.2. ⇑
16 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, 21.1.2. ⇑
17 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, 21.1.2. ⇑
18 Vgl. Wikipedia : Hochleistungsdünnschichtchromatographie, https://de.wikipedia.org/wiki/Hochleistungsd%C3%BCnnschichtchromatographie (zul. gesehen am: 18.06.2021, 13:08) ⇑
19 eigene Abbildung ⇑
20 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, 21.1.2. ⇑
21 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, S. 515. ⇑
22 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, S. 512ff ⇑
23 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, S.513 ⇑
24 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, S. 510 ⇑
25 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, S.517 ⇑
26 eigene Abbildung ⇑
27 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, 21.1.2. ⇑
28 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, 21.1.2. ⇑
29 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, 21.1.2. ⇑
30 Vgl. Deutscher Apotheker-Verlag: Europäisches Arzneibuch, 10. Ausgabe, 1. Nachtrag: Amtliche deutsche Ausgabe (Ph. Eur. 10.1)., München, Deutschland: Deutscher Apotheker Verlag, 2021, 10.0/1617 ⇑
31 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, S.510 ⇑
32 Vgl. Neugebauer, Michael/Rücker, Gerhard/Willems, Günter/Scriba, Gerhard K. E./Hubert, Marcus A.: Instrumentelle pharmazeutische Analytik, 5. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2013, S. 517 ⇑
Monographiebeispiel: Hydrocortisonhydrogensuccinat, Prüfung auf Identität
Stoffcharakterisierung (Wirkung und Anwendung)
Hydrocortisonhydrogensuccinat ist ein Glucocorticoid zur systemischen Anwendung und intravenösen Applikation. Es ist ein weißes bis fast weißes sowie hygroskopisches Pulver, das nicht gut wasserlöslich ist. Dafür ist es löslich in verdünnten Alkalihydroxid- und Alkalicarbonatlösungen sowie leicht löslich in Aceton und wasserfreiem Ethanol. Die Schmelztemperatur liegt bei 169 bis 173°C (je nach Literaturquelle) und der pKS-Wert beträgt 5,1. Außerdem zeigt die Substanz ein Absorptionsmaximum im UV-Spektrum bei 241 nm (gelöst in Ethanol 96%).
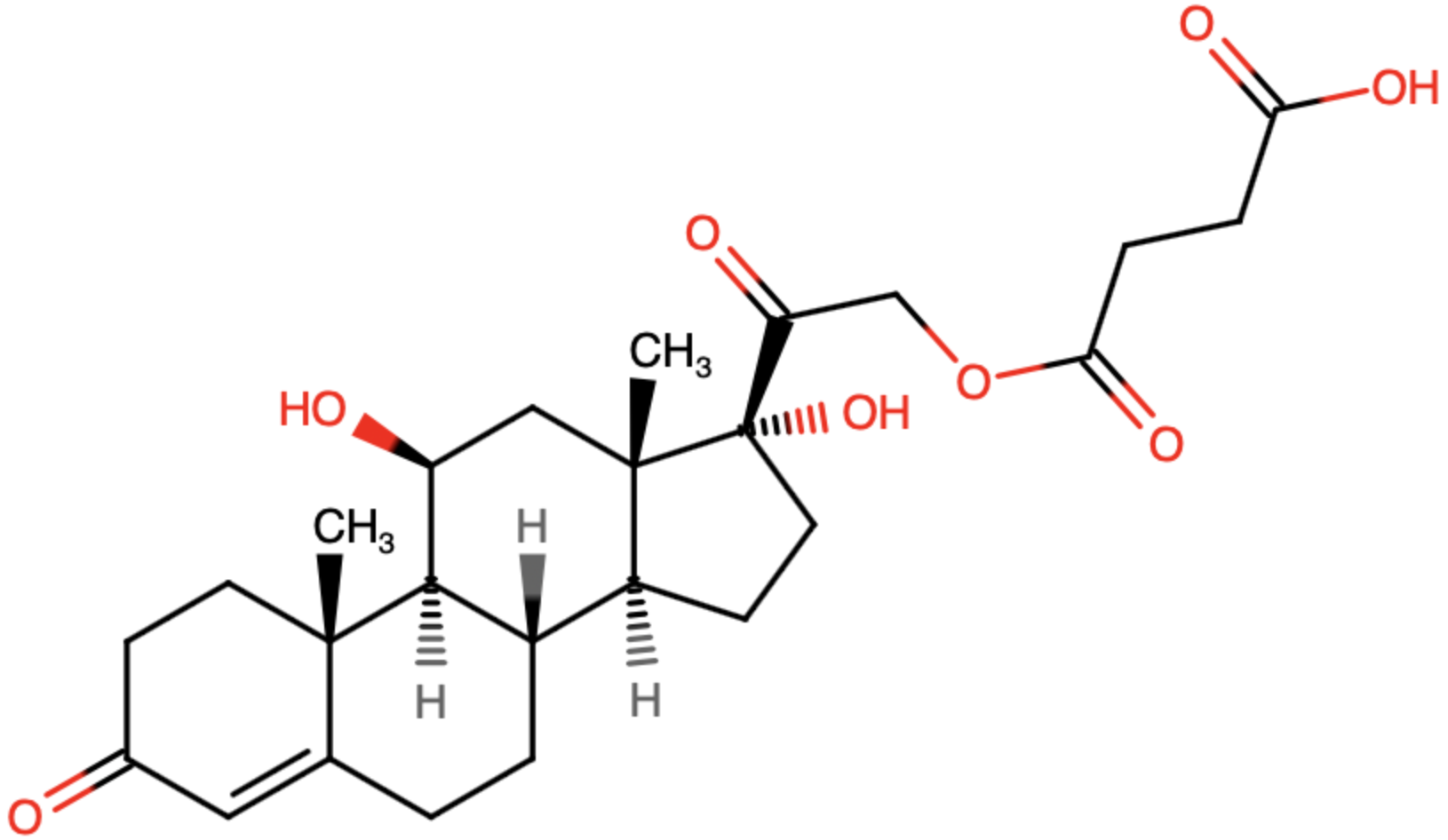
Abbildung 7: Strukturformel von Hydrocortisonhydrogensuccinat (11β,17-Dihydroxy-3,20-dioxopregn-4-en-21-ylhydrogenbutandioat) 1
Es handelt sich um ein hydrophiles Ester-Prodrug, d.h. es wurden die Hydrocortisonmoleküle an der Hydroxygruppe des 21. Kohlenstoffatoms jeweils mit einem Bernsteinsäuremolekül verestert, um so die Wasserlöslichkeit zu erhöhen und eine intravenöse Applikation zu ermöglichen. Diese neu eingeführte Estergruppe wird nach der Applikation wieder durch körpereigene Esterasen gespalten, sodass wieder das rezeptoraktive Hydrocortison (Cortisol) vorliegt.
Da es sich bei dem aktiven Metaboliten um das Nebennierenrindenhormon Cortisol handelt, wird es als Substitutionstherapie bei primären und sekundären Nebenniereninsuffizienzen (Unterfunktion der Nebennierenrinde) sowie bei schweren akuten Schock- oder Stresszuständen, bei denen eine Nebenniereninsuffizienz vorliegt oder vermutet wird, eingesetzt. Ferner wird es auch zur Hemmtherapie bei dem Adrenogenitalen Syndrom (AGS) verwendet.
Die Wirkung von Cortisol beruht auf der Bindung des Cortisols an zytosolische Glucocorticoidrezeptoren und der Beeinflussung der Genexpression durch den Cortisol-Rezeptor-Komplex. Infolgedessen kommt es bei der täglichen Dosierung von 10 bis 30 mg/Tag vor allem zu einer Erhöhung des Blutzuckerspiegels (katabole Wirkung), einer Erhöhung des Fettsäurespiegels (permissive Wirkung), einer Na+-Ionenretention sowie einer K+-Ionensekretion (mineralocorticoide Wirkung), einer verminderten intestinalen Ca2+-Ionenresorption und einer erhöhten renalen Ca2+-Ionenausscheidung (Vitamin-D-antagonistische Wirkung) sowie einer Senkung der Sekretion des Adrenocorticotropen Hormons (ACTH), was eine negative Rückkopplung darstellt. 2 3 4
Durchführung der Monographie
Bei der Prüfung auf Identität gibt es zwei Möglichkeiten diese durchzuführen. Die erste Möglichkeit besteht aus einem infrarotspektroskopischen und einem dünnschicht-chromatographischen Teil.
Für die IR-Spektroskopie wird die Probe und die Referenz (Hydrocortisonhydrogensuccinat CRS) vor der Vermessung bei 100 bis 105°C 3 Stunden lang getrocknet, da die Substanzen hygroskopisch sind.
Für die Dünnschichtchromatographie wird zunächst ein Lösungsmittelgemisch aus einem Volumenteil Methanol R und neun Volumenteilen Dichlormethan R hergestellt. Nun wird die Untersuchungslösung angesetzt, indem man 10 mg der Probensubstanz zu 10 mL des Lösungsmittelgemisches löst. Daraufhin werden zwei Referenzlösungen angemischt. Zum einen eine Referenzlösung a, die aus 20 mg Hydrocortisonhydrogensuccinat CRS besteht, welches zu 20 mL mithilfe des Lösungsmittelgemisches gelöst wird. Zum anderen wird eine Referenzlösung b aus 10 mg Methylprednisolonhydrogensuccinat CRS hergestellt, welches mit der Referenzlösung a zu 10 mL gelöst wird.
Jetzt wird eine feine Bleistiftlinie am unteren Ende einer DC-Platte mit Kieselgel F254 R gezogen und daraufhin noch eine zweite Linie im Abstand von 15 cm (Laufstrecke) von der ersten Linie. Auf die erste Linie werden nun von jeder angesetzten Lösung 5 µL z.B. mithilfe einer kalibrierten Einwegkapillare gegeben und dies in einem Abstand von min. 10 mm zueinander. Die DC-Platte wird nun mit der Auftragungslinie nach unten in eine DC-Kammer gestellt. Diese DC-Kammer wurde zuvor am Boden mit einem Filterpapier bestückt, mit einem Fließmittel aus wasserfreier Ameisensäure R, wasserfreiem Ethanol R und Dichlormethan R im Volumenverhältnis 0,1:1:15 befüllt und verschlossen für ca. eine Stunde bei 20 bis 25°C bis zur Kammersättigung, d.h. bis der gesamte Gasraum der DC-Kammer mit gasförmigem Fließmittel gesättigt ist, stehen gelassen. Die DC-Kammer mit der DC-Platte wird nun vor Licht geschützt und verschlossen gelagert, bis die Laufstrecke von 15 cm erreicht wurde. Danach wird die DC-Platte an der Luft vollständig getrocknet und danach ausgewertet. 5 6
Das Europäische Arzneibuch schlägt noch eine weitere Methode vor, bei der zwei verschiedene Untersuchungslösungen a und b mit jeweils einer dazugehörigen Referenzlösung für die Dünnschichtchromatographie hergestellt werden.
Für die erste Lösung werden 25 mg Substanz in 5 mL Methanol R gelöst, wobei der Vorgang durch Erwärmen beschleunigt wird. Mit Dichlormethan R werden dann 2 mL der so entstandenen Lösung (Stammlösung A) auf 10 mL aufgefüllt. Die Herstellung der dazugehörigen Referenz a erfolgt auf dieselbe Weise unter Verwendung von 25 mg Hydrocortisonhydrogensuccinat CRS. Im Zwischenschritt entsteht hier die Stammlösung B.
Für Untersuchungslösung b werden 2 mL der Stammlösung A in einem 15 mL Reagenzglas mit Schliffstopfen (oder Verschluss aus Polytetrafluorethylen) mit 10 mL einer Lösung von Natriumhydroxid R (0,8 g/L) in Methanol R gemischt. Direkt im Anschluss wird für 5 Minuten Stickstoff R eingeleitet und das Reagenzglas danach verschlossen. Im letzten Schritt wird die Lösung für eine halbe Stunde lichtgeschützt im Wasserbad bei 45 °C stehen und anschließend auf Raumtemperatur abkühlen gelassen.
Analog wird auch hier erneut eine Referenzlösung b hergestellt, wobei die Stammlösung B verwendet wird.
Es werden zwei DC-Platten benötigt, auf die die Untersuchungslösungen mit den jeweiligen Referenzen aufgetragen werden. Als Fließmittel wird eine Mischung von 15 Volumenteilen Ether R und 77 Volumenteilen Dichlormethan R mit einer Mischung von 1,2 Volumenteilen Wasser R und 8 Volumenteilen Methanol R eingesetzt. Die erste der Platten wird nach durchlaufener DC bei 254 nm mit UV-Licht untersucht. Die andere Platte wird zuerst mit ethanolischer Schwefelsäure R besprüht und dann für 10 Minuten oder bis zum Erscheinen von Flecken bei 120°C erhitzt. Nach Erkalten wertet man einmal bei Tageslicht und einmal bei UV-Licht bei einer Wellenlänge von 365 nm aus.
Zusätzlich sieht das Arzneibuch eine Untersuchung auf Fluoreszenz vor, bei der die Substanz in Schwefelsäure R gelöst wird. Die Lösung soll sich braun-rot färben und insbesondere im UV-Licht bei 365nm grün fluoreszieren.
7
Auswertung/ Interpretation/ Bedeutung und Eignung der analytischen Methode
Für die Auswertung der ersten DC-Platte wird diese zunächst unter UV-Licht bei 254 nm bestrahlt und begutachtet, ob der Hauptfleck der Untersuchungslösung in Bezug auf Lage und Größe dem Hauptfleck der Referenzlösung a entspricht. Dabei fluoresziert die DC-Platte aufgrund eines Fluoreszenzindikators, wohingegen die Flecken nicht fluoreszieren und dunkel erscheinen. Die Lage wird mithilfe des Retentionsfaktors Rf beschrieben (siehe Abbildung 8):
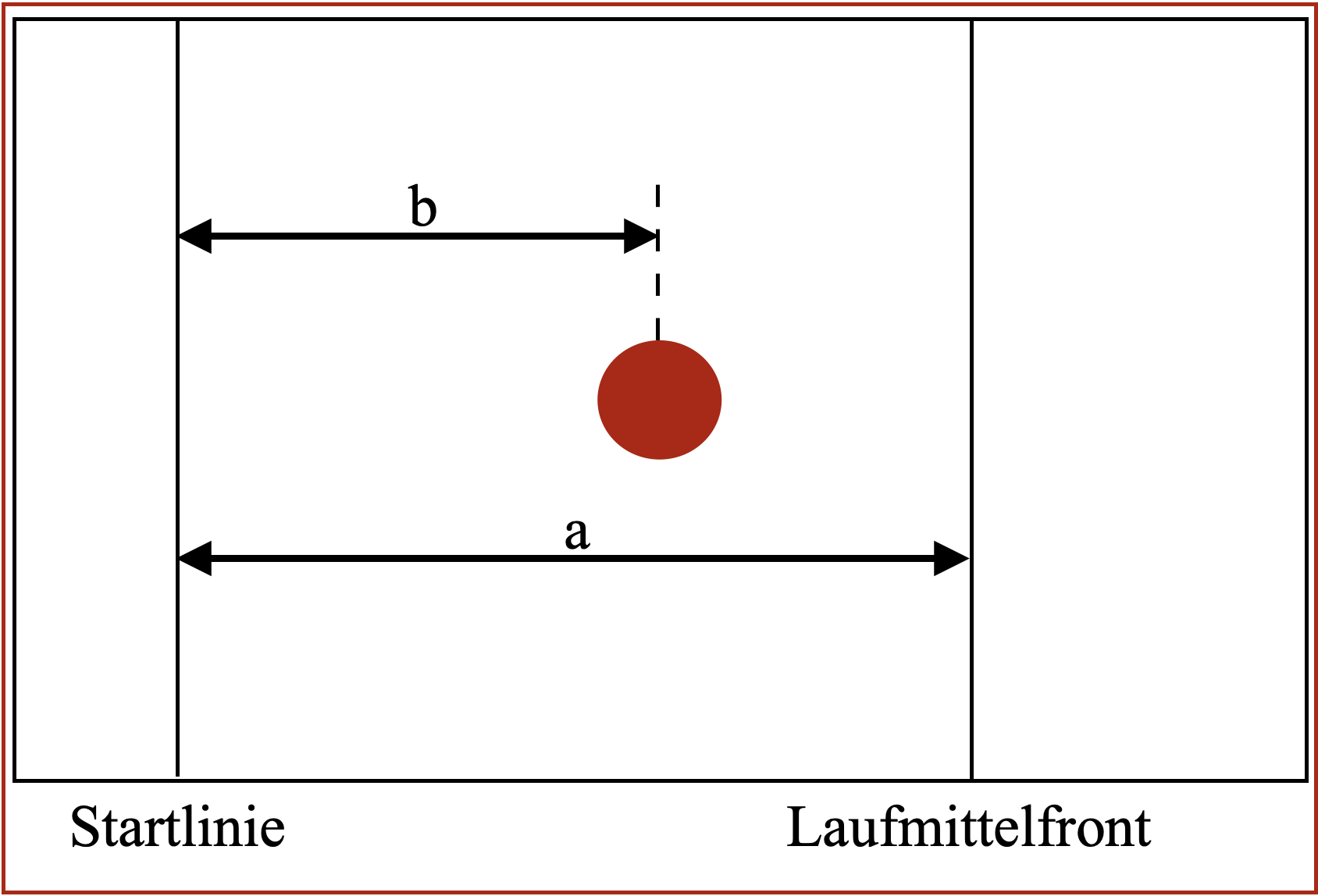
⚠ $$ R_{f} = {b\over a} ⚠ $$
Nun wird die DC-Platte mit ethanolischer Schwefelsäure R besprüht, für 10 min. oder bis zum Erscheinen von Flecken bei 120°C erhitzt und daraufhin erkalten gelassen. Infolgedessen wird der Hauptfleck der Untersuchungslösung mit dem Hauptfleck der Referenzlösung a in Bezug auf Lage und Farbe im Tageslicht und unter UV-Bestrahlung bei 365 nm (Flecken fluoreszieren) verglichen. Stimmen nun alle Parameter überein, so entspricht die Probe bei passendem IR-Spektrum den Anforderungen des Europäischen Arzneibuchs.
Um die Eignung dieser Methode der Dünnschichtchromatographie zu prüfen, wird die Trennleistung beurteilt, indem man das Ergebnis der Referenzlösung b betrachtet. Diese sollte 2 Flecken zeigen, die eventuell nicht vollständig voneinander getrennt sind. 9 10
Bei der zweiten Methode werden auf der ersten DC-Platte die Hauptflecken, die durch die Untersuchungslösungen entstanden sind, mit denen der Referenzlösung hinsichtlich der Lage und der Größe verglichen und sollten einander entsprechen.
Die zweite Platte wird außer der Lage und Größe der Hauptflecken zusätzlich auf Farbe im Tageslicht und Fluoreszenz im UV-Licht bei 365 nm untersucht. Dabei sollen auch hier die Flecken der Untersuchungslösungen den Flecken der Referenzlösungen entsprechen. Untersuchungslösung b sowie die dazugehörige Referenzlösung b weisen einen wesentlich größeren Rf-Wert auf als die Lösungen a.
Diese Methode eignet sich für eine Identitätsprüfung eher schlecht. Zum einen ist sie zeit- und arbeitsaufwendig und wäre durch die HPLC, die das Arzneibuch für die Reinheitsprüfung von Hydrocortisonhydrogensuccinat vorsieht, redundant. Ebenso ist sie weniger für den Apothekenmaßstab angedacht, da der verwendete Stickstoff in den wenigsten Fällen in einem Apothekenlabor verfügbar ist. 11 12
Einzelnachweise
1 eigene Abbildung ⇑
2 Vgl. Geisslinger, Gerd/Sabine Menzel/Thomas Gudermann/Burkhard Hinz/Peter Ruth/Mutschler: Mutschler Arzneimittelwirkungen: Pharmakologie - Klinische Pharmakologie - Toxikologie, Baden-Baden, Deutschland: Nomos Verlagsgesellschaft, 2019, S. 730-743. ⇑
3 Vgl. Hydrocortison®: in: Pfizer.de, o. D., https://www.pfizer.de/medikamente-patientenhilfe/medikamente/hydrocortisonR (abgerufen am 18.05.2021 um 12:00 Uhr). ⇑
4 Vgl. Bracher, Franz/Peter Heisig/Peter Langguth/Ernst Mutschler/Tanja Schirmeister/Gerhard Scriba/Elisabeth Stahl-Biskup/Reinhard Troschütz: Arzneibuch-Kommentar Online VOL 66: Wissenschaftliche Erläuterungen zum Arzneibuch, 1. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2021, 4.00/0768. ⇑
5 Vgl. Deutscher Apotheker-Verlag: Europäisches Arzneibuch, 10. Ausgabe, 1. Nachtrag: Amtliche deutsche Ausgabe (Ph. Eur. 10.1)., München, Deutschland: Deutscher Apotheker Verlag, 2021, 10.0/2.02.2700, 10.0/0768. ⇑
6 Vgl. Bracher, Franz/Peter Heisig/Peter Langguth/Ernst Mutschler/Tanja Schirmeister/Gerhard Scriba/Elisabeth Stahl-Biskup/Reinhard Troschütz: Arzneibuch-Kommentar Online VOL 66: Wissenschaftliche Erläuterungen zum Arzneibuch, 1. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2021, 4.00/0768. ⇑
7 Vgl. Deutscher Apotheker-Verlag: Europäisches Arzneibuch, 10. Ausgabe, 1. Nachtrag: Amtliche deutsche Ausgabe (Ph. Eur. 10.1)., München, Deutschland: Deutscher Apotheker Verlag, 2021, 10.0/2.02.2700, 10.0/0768. ⇑
8 In Anlehnung an Kommentar zu Ph. Eur. 9.0/2.02.27.00 Dünnschichtchromatographie ⇑
9 Vgl. Deutscher Apotheker-Verlag: Europäisches Arzneibuch, 10. Ausgabe, 1. Nachtrag: Amtliche deutsche Ausgabe (Ph. Eur. 10.1)., München, Deutschland: Deutscher Apotheker Verlag, 2021, 10.0/0768. ⇑
10 Vgl. Bracher, Franz/Peter Heisig/Peter Langguth/Ernst Mutschler/Tanja Schirmeister/Gerhard Scriba/Elisabeth Stahl-Biskup/Reinhard Troschütz: Arzneibuch-Kommentar Online VOL 66: Wissenschaftliche Erläuterungen zum Arzneibuch, 1. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2021, 4.00/0768. ⇑
11 Vgl. Deutscher Apotheker-Verlag: Europäisches Arzneibuch, 10. Ausgabe, 1. Nachtrag: Amtliche deutsche Ausgabe (Ph. Eur. 10.1)., München, Deutschland: Deutscher Apotheker Verlag, 2021, 10.0/2.02.2700, 10.0/0768. ⇑
12 Vgl. Bracher, Franz/Peter Heisig/Peter Langguth/Ernst Mutschler/Tanja Schirmeister/Gerhard Scriba/Elisabeth Stahl-Biskup/Reinhard Troschütz: Arzneibuch-Kommentar Online VOL 66: Wissenschaftliche Erläuterungen zum Arzneibuch, 1. Aufl., Stuttgart, Deutschland: Wissenschaftliche Verlagsgesellschaft, 2021, 4.00/0768. ⇑
TU-Braunschweig Institut für Medizinische und Pharmazeutische Chemie Seminar: Instrumentelle Analytik Kontakt: tubs@t-kellner.de
