Atomemissionsspektroskopie
Titelblatt
Bericht der Expertengruppe für
Atomemissionsspektroskopie
SoSe 2021
Abgabedatum
07.06.2021
Über-/Expertengruppe 09
Eda Nur Abaci
Regina Körössy
Bibulat Idris
Muhammed Kamil Asi
Yousef Mahrougui
Raphael Tahan
Atomemissionsspektroskopie
Inhaltsverzeichnis
Einleitung
Die Atomemissionsspektrometrie (AES) ist ein Teilbereich der Atomspektrometrie und beschäftigt sich mit dem Aussenden von elektromagnetischer Strahlung durch einen Analyten. Sie wird heutzutage auch als optische Emissionsspektrometrie (OES) bezeichnet. Im Gegensatz zur Atomabsorptionsspektrometrie (AAS) handelt es sich hierbei um die Messung der Emission von Atomen. Anwendungsgebiete sind beispielsweise die quantitative und qualitative Analytik, wobei die Flammenphotometrie beziehungsweise Flammenspektrometrie als quantitative Methode die am häufigsten angewendete Form darstellt. Hierbei wird die Analysensubstanz mit einer meist wässrigen Lösung in die Flamme gesprüht und die Intensität von Linien in einem Linienspektrum ausgewertet. Vor allem Elemente, deren Emissionslinien im sichtbaren Bereich des Spektrums eine hohe Intensität haben, lassen sich mit dieser Methode problemlos nachweisen. Hierzu zählen vor allem die Salze von Alkali- und Erdalkalimetallen. Es ist in der Regel nicht von Bedeutung, ob das nachzuweisende Element in ionischer oder kovalenter Form in einem Molekül gebunden vorliegt. Im Wesentlichen ist die Flammenphotometrie sowohl von der Eigenemission der Flamme, als auch von ihrer Temperatur und Geometrie abhängig. Ein weiteres Verfahren ist die ICP-OES (Inductively coupled plasma optical emission spectrometry), welche neben der Spuren- und Wasseranalytik, mittlerweile auch bei der Analyse von radioaktiven Substanzen ihren Einsatz findet, da lediglich das emittierte Licht analysiert wird und die Radioaktivität keine Störung darstellt. 1 2 3
Physikalische und chemische Grundlagen
Prinzip
Das Prinzip der Atomemissionsspektroskopie beruht darauf, dass durch Zufuhr von Energie Valenzelektronen von einem energieärmeren Grundzustand auf ein höheres, zuvor nicht besetztes Energieniveau angehoben werden. Dieser angeregte Zustand ist instabil, wodurch Elektronen das höhere Energieniveau nicht dauerhaft besetzen können und in kürzester Zeit auf ihr Ausgangsenergieniveau zurückfallen. Die freiwerdende Energie wird in Form von Lichtquanten emittiert. Die Frequenz beziehungsweise die Wellenlänge der freiwerdenden Quanten entspricht genau der Energiedifferenz der beiden überkreuzten Energieniveaus.
⚠ $$ ∆𝐸 = 𝐸_{2} − 𝐸_{0} = h \cdot 𝑣 = \left(\frac{h\cdot 𝑐}{λ}\right) ⚠ $$
- ∆𝐸: Energiedifferenz
- 𝐸2: Energie des angeregten Zustands
- 𝐸0: Energie Grundzustand
- h: Plancksches Wirkungsquantum [Js]
- 𝑣: Frequenz
- 𝑐: Phasengeschwindigkeit
- λ: Wellenlänge
Somit entstehen durch Energieaufnahme und -abgabe Atomspektren, die für jedes Atom spezifisch und charakteristisch sind.
Prinzipiell können in einem Atom mehrere Übergänge angeregt werden, wodurch Linienspektren entstehen. Atome können die Energie, die sie durch die Anregung erhalten haben, jedoch nur durch das Zurückfallen und nicht wie Moleküle durch Rotation oder Schwingung abgeben. Grundsätzlich können Atome auf verschiedene Art und Weise angeregt werden, wie in Form von Lichtenergie oder thermischer Energie, wobei Letzteres in der Atomemissionsspektroskopie genutzt wird. 4
Ionisierung
Die Energiezufuhr muss für die Atomisierung, also für die Bindungsspaltung und für das Anregen der Atome ausreichen, darf aber nicht zu hoch sein, da sonst die Ionisierungsenergie überschritten werden kann. Bei der Ionisierung verliert ein elektrisch neutrales Atom ein oder mehrere Elektronen, dies führt zu eigenen Ionenspektren. Aufgrund der Interferenzen und weiterer Linien im Spektrum die entstehen, ist die Ionisierung grundsätzlich unerwünscht. Das für die Auswertung notwendige Gesamtbild könnte nämlich ungenau werden, was sowohl die exakte quantitative als auch die qualitative Auswertung dieses Verfahrens unmöglich macht. 5
Instrumenteller Aufbau
Allgemeiner Aufbau
Allgemein lässt sich der instrumentelle Aufbau eines Atomemissionsspektrometers in vier wesentliche Komponenten unterteilen: Zerstäuber/Probenzufuhr, Atomisator, Monochromator und Detektor. Je nach Wahl des Atomisators kann dabei unterschieden werden zwischen Flammen-Atomemissionsspektrometrie und Plasma-Emissionsspektrometrie. In beiden Methoden wird dabei durch einen Detektor die Lichtemission einer Analysensubstanz gemessen, nachdem die Atome durch hohe Temperaturen angeregt wurden. 6
Flammen-Atomemissionsspektrometrie (F-AES)
Allgemein
In der F-AES wird die Probe zunächst mithilfe von Pressluft über eine Kapillare zerstäubt (s. Abb.: 1) ). Grobe Tröpfchen aus der Zerstäubung kondensieren dabei wieder zurück in das Probengefäß. Nach Verlassen der Zerstäuberkammer wird die zerstäubte Lösung (Aerosol) mit Brenngas vermischt und anschließend zum Brenner geleitet (s. Abb.: 2) ). Nach Eintritt in die Flamme wird die Lichtemission der Atome durch einen Hohlspiegel zum Monochromator reflektiert (s. Abb.: 3) ). Mithilfe des Monochromators werden lediglich die gewünschten Spektrallinien erfasst und zum Detektor geleitet. Im Detektor wird das Licht, welches von dem Monochromator gefiltert wurde, in ein elektrisches Signal umgewandelt und abschließend auf dem Gerät angezeigt (s. Abb.: 4) ). 7
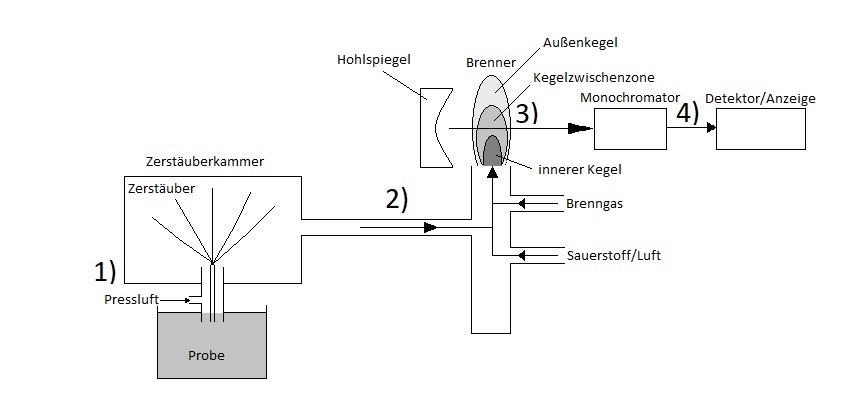
Flammentemperatur
Die Flammentemperatur ist ein überaus wichtiger Parameter der F-AES, da sie entscheidend für die Atomisierung ist. Mit zunehmender Temperatur steigt nämlich die Zahl der angeregten Atome und demzufolge auch die Lichtemission bzw. Lichtintensität, indem die Atome wieder in den Grundzustand fallen. Diese Gesetzmäßigkeit wird dabei durch die Boltzmann-Gleichung beschrieben:
⚠ $$ \left(\frac{N^*}{N_{0}}\right) = g \cdot e^\left(\frac{-∆E}{k \cdot T}\right) ⚠ $$
- N*: Zahl der angeregten Atome
- N0: Zahl der Atome im Grundzustand
- g: statistischer Faktor
- ΔE: Anregungsenergie
- k: Boltzmann-Konstante [J/K]
- T: Temperatur [K]
Durch den Anstieg der Temperatur wird jedoch auch die Ionisierung von Atomen begünstigt, welche für die Auswertung unerwünscht sind, da nur die angeregten Atome zur Lichtemission führen. Es kann dabei aber eine so große Temperatur gewählt werden, dass auch die Ionen in einen angeregten Zustand gebracht werden und somit auch zur Lichtemission beitragen. Die erreichten Temperaturen in der Flamme sind dabei abhängig von dem Brenngasgemisch und der Geometrie der Flamme. Das Brenngasgemisch besteht dabei immer aus einem Brenngas und einem Oxidationsmittel. Folgende Kombinationen sind hier in Abhängigkeit von der Analyse möglich:
| Brenngas + Oxidationsmittel | Maximale Temperaturen [Celsius] |
| Acetylen + Luft | 2100-2400 |
| Acetylen + Sauerstoff | 3000-3150 |
| Propan + Luft | 1700-1900 |
| Propan + Sauerstoff | 2700-2800 |
| Wasserstoff + Luft | 2000-2150 |
| Wasserstoff + Sauerstoff | 2500-2700 |
Aufgrund der Geometrie der Flamme herrschen Temperaturschwankungen in der Flamme. Maximale Temperaturen werden dabei vor allem in der Kegelzwischenzone oberhalb der primären Verbrennungszone des Flammenkegels erreicht (s. Abb.: Aufbau einer Flammenemissionsspektrometrie). Außerdem muss während der Atomisierung eine ausreichende Flammenstabilität gewährleistet sein. In Anbetracht dieser variablen Parameter sollte vor einer quantitativen Analyse eine Kalibrierung durchgeführt werden. 9
Vorgänge in der Flammentechnik
Mithilfe der Flammentechnik sollen die Atome der Probelösung in einen gasförmigen Zustand gebracht werden, um schließlich angeregt zu werden. Dafür muss die Analysenlösung mehrere chemisch-physikalische Zustände durchlaufen. Zunächst bildet sich also ein Aerosol aus Brenngasgemisch und Probe (fest, z.B. NaCl). Dabei werden Lösemittel und ggf. Oxide entfernt. Im nächsten Schritt werden die Moleküle im Gasgemisch verflüssigt und schließlich durch Dissoziation in einen gasförmigen Zustand aus Atomen überführt. Diese Atome können dann durch die Temperaturen der Flamme angeregt werden. Während des Atomisierungsprozesses können neben den gewünschten angeregten Atomen auch unerwünschte Teilchen, wie z.B. Ionen, Radikale oder angeregte Moleküle entstehen. 10
Plasma-Emissionsspektrometrie (ICP-OES)
Prinzip
In der ICP-OES (Inductively coupled plasma optical emission spectrometry) werden Plasmen zur Anregung von Elektronen verwendet, welche im Vergleich zur Flammentechnik noch höhere Temperaturen erzeugen können. In diesem Verfahren werden die Atome durch die hohen Temperaturen ionisiert, sodass am Ende ein Ionenspektrum ausgewertet wird. Die dabei verwendete ICP-Einheit besteht aus einem Zerstäuber, einem Hochfrequenz-Generator und einem Plasmabrenner (s. Abb.).
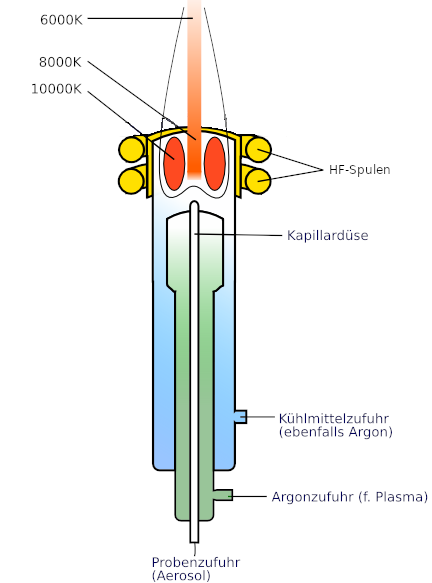
Bevor die Probe das Plasma betritt muss diese zu einem Aerosol mit möglichst feinen Tröpfchen zerstäubt werden, da andernfalls das Plasma erloschen werden kann. Ein Plasma wird definiert als ein elektrisch leitendes und gasförmiges System, in dem sich Atome, Moleküle, positive und negative Ionen wie auch Elektronen befinden. Besonders die geladenen Teilchen sind aufgrund der Übertragung ihrer Bewegungsenergie wichtig für das Erreichen hoher Temperaturen. Zur Erzeugung eines Plasmas muss elektrische Energie auf ein Gas (meistens Argon) übertragen werden, wodurch das Gas ionisiert wird.
In der ICP-OES erfolgt diese Ionisierung von Argon mithilfe einer Induktionsspule eines Hochfrequenz-Generators (HF-Spule), welche um drei konzentrische Quarzrohre, dem Plasmabrenner, gelegt ist. Im inneren Quarzrohr (Abb.: Kapillardüse) liegt das Proben-Aerosol vor, welches mit dem Trägergas Argon aus dem mittleren Rohr (Abb.: grün) in das Argon-Plasma im äußeren Rohr eindringt (Abb.: blau), wo die Probe dann wiederum erhitzt wird. Das dabei emittierte Licht wird durch einen Monochromator in seine Wellenlängen zerlegt, um dann mithilfe der ermittelten Wellenlängen bzw. der Lichtintensität qualitative und quantitative Aussagen zu treffen. 12
Vor- und Nachteile
Durch die hohen Temperaturen des Plasmas (bis zu 10000 K; Vergleich: F-AES nur bis ca. 3000 K) werden viele störende Bestandteile der Probe entfernt, um letztendlich nur die Atome des Analyten durch Anregung zu erfassen. Im Gegensatz zur F-AES werden die Atome in der ICP-OES aufgrund der hohen Temperaturen bewusst ionisiert, da in diesem Verfahren die Ionenspektren betrachtet werden, welche im Gegensatz zu den Atomspektren der F-AES weniger störanfällig sind. Die ICP-OES weist außerdem auch eine niedrigere Nachweisgrenze auf, da das Verfahren mit deutlich homogeneren Temperaturen in der Plasmafackel arbeitet. Dadurch hat sich die Methode besonders in der pharmazeutischen Industrie bewährt, in der sie oftmals durch Kopplung mit z.B. der HPLC noch niedrigere Nachweisgrenzen von bis zu 10ppt ermöglicht und zudem auch noch Multielementanalysen durchführen kann.
Nachteile der ICP-OES sind u.a. die hohen Kosten von Argon, die langen Analysezeiten und die spektralen Interferenzen, welche entweder durch Begleitsubstanzen (Wasser, Stickstoff) und/oder durch Eigenstrahlung des Plasmas sogenannte "Begleitspektren" bilden können. Die spektralen Interferenzen lassen sich durch Monochromatoren mit einer hohen Auflösung vermeiden. 13 14
Einzelnachweise
1 Vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 77-80 ⇑
2 Vgl. Instrumentelle Analytik kompakt / Dominik, Andreas (2013), S. 55-58 ⇑
3 Vgl. https://www.spektrum.de/lexikon/chemie/atomemissionsspektrometrie/788 , als letztes abgerufen: 07.06.2021 ⇑
4 Vgl. https://www.chemie.de/lexikon/Atomspektroskopie.html , als letztes abgerufen: 21.06.2021, 22:02 Uhr ⇑
5 Vgl. https://www.chemie.de/lexikon/Atomspektroskopie.html , als letztes abgerufen: 21.06.2021, 22:02 Uhr ⇑
6 Vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 79 f. ⇑
7 Vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 77-80 ⇑
8 Eigene Zeichnung, in Anlehnung an: Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 79 ⇑
9 Vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 77-80 ⇑
10 Vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 77-80 ⇑
11 Veränderte Abbildung: Vgl. https://de.wikipedia.org/wiki/Massenspektrometrie_mit_induktiv_gekoppeltem_Plasma , als letztes abgerufen: 21.06.2021, 21:22 Uhr ⇑
12 Vgl. Analytische Chemie: Grundlagen, Methoden und Praxis / Georg Schwedt, Torsten C. Schmidt und Oliver J. Schmitz, S. 248-250 ⇑
13 Vgl. http://dodo.fb06.fh-muenchen.de/maier/analytik/Blaetter/N101_Emission_Fluorimetrie1_a_BAneu.pdf , als letztes abgerufen: 19.06.2021, 22:32 Uhr ⇑
14 Vgl. Analytische Chemie: Grundlagen, Methoden und Praxis / Georg Schwedt, Torsten C. Schmidt und Oliver J. Schmitz, S. 248-250 ⇑
Monographiebeispiel: Calciumacetat (Prüfung auf Reinheit, Gehalt an Kalium)
Stoffcharakterisierung (Wirkung und Anwendung)
Calciumacetat wirkt als Phosphatbinder und bindet Phosphat aus der Nahrung. Es wird eingenommen, um einer Erhöhung des Phosphatspiegels im Blut (Hyperphosphatämie) entgegenzuwirken. Calciumacetat wird auch bei chronischer Nierenschwäche mit angezeigter Dialyse angewendet. 1
Durchführung der Monographie
In der Monographie von Calciumacetat schreibt das Arzneibuch zur Bestimmung des Kaliumgehalts eine Reinheitsprüfung mithilfe der Atomemissionsspektrometrie vor, genauer mithilfe der Flammenphotometrie. Der Gehalt an Kalium darf dabei, wenn die Substanz zur Herstellung von Parenteralia oder Hämodialyselösungen bestimmt ist, 500 ppm 2 nicht überschreiten. Die Reinheitsprüfung soll mithilfe der Standardadditionsmethode (Methode II) 3 durchgeführt werden. Die Messungen der Emissionsintensitäten erfolgen bei einer Wellenlänge von 766,5 nm. 4
Die Untersuchungslösung wird hergestellt, indem 1,00 g Substanz in Wasser R zu 25,0 mL gelöst wird. In mindestens drei gleich großen Messkolben werden gleiche Volumina der Untersuchungslösung hinzugefügt. Bis auf einen Messkolben werden in die anderen Messkolben steigende Volumina der Kalium-Lösung (0,2 % K) R hinzugegeben. Alle Messkolben werden mit Wasser R auf ein gleiches Volumen aufgefüllt. So werden eine Probenlösung, deren Gehalt an Kalium ermittelt werden soll, und eine Reihe von Referenzlösungen mit steigenden Kalium-Konzentrationen erhalten. Zur Bestimmung eines geeigneten Messbereichs wird der Nullpunkt mit Wasser R und der Vollausschlag mit der am höchsten konzentrierten Referenzlösung bestimmt. Anschließend wird jede Lösung in das Gerät eingebracht und jeweils mehrfach gemessen, wobei für jede Lösung die gleiche Anzahl an Messungen durchgeführt wird. 5 6
Auswertung/ Interpretation/ Bedeutung und Eignung der analytischen Methode
Auswertung und Interpretation
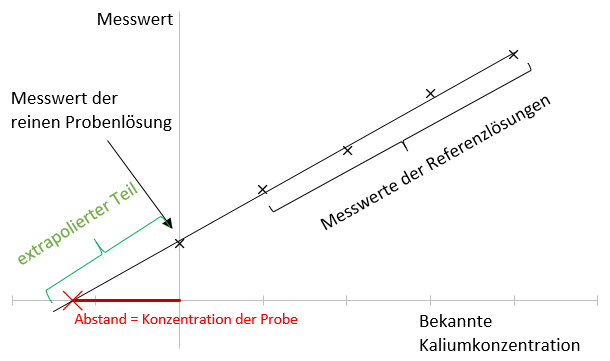
Die Mittelwerte der erhaltenen Messwerte der jeweiligen Lösungen werden grafisch in einem Koordinatensystem aufgetragen. Auf der Abszisse werden die bekannten Kalium-Konzentrationen der Lösungen aufgetragen, auf der Ordinate die jeweiligen Mittelwerte der Messwerte. Der Mittelwert der Messwerte der reinen Probenlösung wird beim Nullpunkt der Abszisse aufgetragen. Nachdem die Punkte im Koordinatensystem aufgetragen wurden, wird eine Gerade nach der Methode der kleinsten Fehlerquadrate für diese Punkte ermittelt, welche mit einer Funktionsgleichung der Form ⚠ $y=mx+b$ beschrieben werden kann.7 Um die Steigung dieser Geraden zu berechnen, wird folgende Formel herangezogen:
⚠ $$ m = \frac{\sum\nolimits_{i=1}^{n} (x_i - \overline x) \cdot (y_i - \overline y)}{\sum\nolimits_{i=1}^{n} (x_i - \overline x)^2}⚠ $$
Der Schnittpunkt der Geraden mit der Ordinate wird mit folgender Formel berechnet:
⚠ $$ b = \overline y - m \cdot \overline x ⚠ $$
Hierbei stehen die einzelnen Formelzeichen für folgende Größen:
⚠ $x_i$= Konzentrationswerte⚠ $\overline x$= Mittelwert der Konzentrationen⚠ $y_i$= Messwerte⚠ $\overline y$= Mittelwert der Messwerte⚠ $n$= Anzahl der vermessenen Lösungen 9
Mithilfe der ermittelten Funktionsgleichung kann die entsprechende Ausgleichsgerade eingezeichnet werden. Anschließend kann durch Extrapolation der Schnittpunkt dieser Geraden mit der Abszisse bestimmt werden. Die unbekannte Konzentration der Probe kann ermittelt werden, indem der Abstand vom Nullpunkt bis zu jenem Schnittpunkt bestimmt wird.10 Zur Bestimmung der Probenkonzentration kann auch die ermittelte Funktionsgleichung gleich Null gesetzt und anschließend gelöst werden, wobei der Betrag des ermittelten x-Werts verwendet wird. Mithilfe dieser Konzentration kann unter Beachtung der Verdünnungsfaktoren die Masse an Kalium berechnet werden, die sich in dem einen Gramm Calciumacetat-Analysesubstanz befand. Diese Masse an Kalium wird durch die Gesamtmasse, also 1,00 g Analysesubstanz, dividiert, wodurch der Massenanteil von Kalium in der Analysensubstanz erhalten wird. Dieser darf nicht größer als 500 ppm sein.11
Bedeutung und Eignung
Die Methode eignet sich deshalb für die Reinheitsprüfung, da sie eine niedrige Bestimmungsgrenze hat. So können bereits Mengen von ca. 0,05 µg/mL an Kalium bestimmt werden.12 Zudem gehört Kalium zu den Alkalimetallen, welche sich dadurch auszeichnen, dass sie bereits bei relativ geringen Temperaturen angeregt werden können.13 Dies hat zur Folge, dass Kalium dementsprechend eine höhere Lichtemissionsintensität aufweist, da mehr Atome bereits bei niedrigeren Temperaturen angeregt werden und somit mehr Atome Licht emittieren (Boltzmann-Gleichung).14 Daraus resultiert letztendlich, dass eine gute quantitative Bestimmung mithilfe der Flammenphotometrie möglich ist.
Einzelnachweise
1 Vgl. https://image.wub-service.de/resources/static/des/210515/29/86/29860.pdf , als letztes abgerufen: 01.06.2021, 9:48 Uhr ⇑
2 Vgl. Ph. Eur. 10.0/ 2128 ⇑
3 Vgl. Ph. Eur. 10.0/ 2.02.22.00 ⇑
4 Vgl. Ph. Eur. 10.0/ 2128 ⇑
5 Vgl. Ph. Eur. 10.0/ 2.02.22.00 ⇑
6 Vgl. Arzneibuch-Kommentar 6.0/ 2.02.23.00 ⇑
7 Vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 83 ⇑
8 Selbst erstellte Abbildung ⇑
9 Vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 28 f. ⇑
10 Vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 83 ⇑
11 Vgl. Ph. Eur. 10.0/ 2128 ⇑
12 Vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 81 ⇑
13 Vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 78 ⇑
14 Vgl. Rücker, G., Neugebauer, M. & Willems, G. G.(2013). Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 78 f. ⇑
TU-Braunschweig Institut für Medizinische und Pharmazeutische Chemie Seminar: Instrumentelle Analytik Kontakt: tubs@t-kellner.de
