Atomabsorptionsspektroskopie
Bericht der Expertengruppe für
Atomabsorptionsspektroskopie
SoSe 2021
Über-/Expertengruppe 10
Lena Rethmeyer
Maja Dominique Voßgröne
Klara Hofmann
Lilli Arnold
Meret Krull
Charlotte Willuhn
Atomabsorptionsspektroskopie
Inhaltsverzeichnis
Einleitung
Die Atomabsorptionsspektroskopie (AAS) ist eine absorptionsspektroskopische Methode der pharmazeutischen Analytik zur quantitativen und qualitativen Analyse eines chemischen Elements in einer Probe.
Alan Walsh, geboren am 19. Dezember 1916, gilt als Entwickler der Atomabsorptionsmethode der chemischen Analyse und hat damit die quantitative Analyse bereits in den 1950er Jahren revolutioniert.
Die schnelle und hochempfindliche Methode dient der Bestimmung der Konzentrationen von mehr als 65 Elementen (Metalle, Halbmetalle) mit niedriger Nachweisgrenze von oft unter 1 ppm. Herkömmliche nasschemische Methoden wurden somit überflüssig. Die AAS findet weltweit Anwendung in vielen unterschiedlichen Gebieten wie der Medizin, Landwirtschaft, Mineralexploration, Metallurgie, Lebensmittelanalyse, Biochemie, Umweltkontrolle und der Pharmazie. Letzteres impliziert vor allem Grenzwert- und Reinheitsprüfungen auf anorganische Verunreinigungen und organometallischen Verbindungen.1 2
Physikalische und chemische Grundlagen
Die AAS misst, wie stark Licht, meist im UV/VIS-Bereich, von Atomen absorbiert wird.
Dafür wird eine Lösung von Metallsalzen zunächst in einer Flamme verdampft: Die Atome werden durch Einsprühen einer Lösung der zu untersuchenden Probe in einer Gasbrennerflamme getrennt. Bei dieser sogenannten Atomisierung werden alle chemischen Bindungen gespalten und es wird für element-typische Anregungsenergien der Elektronen gesorgt.
Es folgt die Resonanzanregung, bei der die gebildeten Atome mit Licht geeigneter Wellenlänge bestrahlt und die Elektronen dadurch in einen angeregten Zustand versetzt werden. Zur Anregung der Atome wird die Emissionslinie des gleichen Elements verwendet, welches analysiert werden soll. So wird der gleiche Elektronenübergang der Atome in der Probe angeregt, dem die Linie entstammt. Diese Linie wird als Resonanzlinie bezeichnet und der Vorgang somit als Resonanzabsorbtion. Für die Messungen muss gelten, dass die Messwellenlänge gleich der Absorptionswellenlänge ist, andernfalls können die Atome die Strahlung nicht absorbieren. Das Spektrum von Atomen ist ein Linienspektrum, da als Anregung nur die Elektronenanregung in höher liegende Orbitale stattfindet. Die Absorptionslinien lassen sich aufgrund von Anregungsmöglichkeiten in alle Orbitale zu Serien zusammenfassen, die alle vom selben Ausgangszustand ausgehen.3
Beim Durchgang durch die Probe wird die Intensität des eingestrahlten Lichtes um den Betrag der Absorption vermindert. Die dabei absorbierte Lichtintensität wird gemessen und dieser Messwert zur Quantifizierung der Analyse genutzt. Die Elektronen angeregter Atome fallen nachfolgend in den Ausgangszustand zurück, wobei die überschüssige Energie als Strahlung wieder abgegeben wird. Die Atome emittieren dabei Licht mit genau der Wellenlänge, die zur Anregung absorbiert wurde. Dies nennt man Emissionsspektrum der Atome. Es ist trotzdem möglich, die Intensitätsschwächung als Absorptionssignal zu messen, da das eingestrahlte Licht durch die Optik des Messgerätes auf einen kleinen Raumwinkel fokussiert wird. Im Gegensatz dazu erfolgt die Emission des Lichts durch die Probe als Kugelwelle über den gesamten Raum. Nur ein vernachlässigbar kleiner Anteil davon gelangt mit dem Licht der Lampe durch den Austrittsspalt.4 5
Ausgewertet werden die Messungen mithilfe des Lambert-Beer'schen Gesetzes.
⚠ $\frac{I_0}{I} )$ = 𝜺𝝺 ∙ c ∙ d I0 : Intensität des eingestrahlten Lichtes (Einheit: W·m−2)
I : Intensität des transmittierten Lichtes (Einheit: W·m−2)
𝜺𝝺 : Absorptionskoeffizient (Einheit L·mol-1·cm-1)
d : Schichtdicke des durchstrahlten Körpers (Einheit: m)
c : Stoffmengenkonzentration der absorbierenden Substanz in der Flüssigkeit (Einheit: mol·m−3)
Es gilt, dass die logarithmisch definierte Absorption proportional zur Konzentration der Probe ist. Steigt die Konzentration der analysierten Substanz in der Probe, steigt auch die Schwächung des eingestrahlen Lichts proportional an. In der Praxis allerdings gibt es jedoch keinen direkten linearen Zusammenhang. Außerdem sind weder die Konzentration c noch die Schichtdicke d bekannt. Der molare Absorptionskoeffizient 𝜺(𝝺) lässt sich demzufolge nicht berechnen, weshalb die Auswertung mithilfe einer Kalibriergerade erfolgt.6 7
Instrumenteller Aufbau
Prinzipieller Aufbau
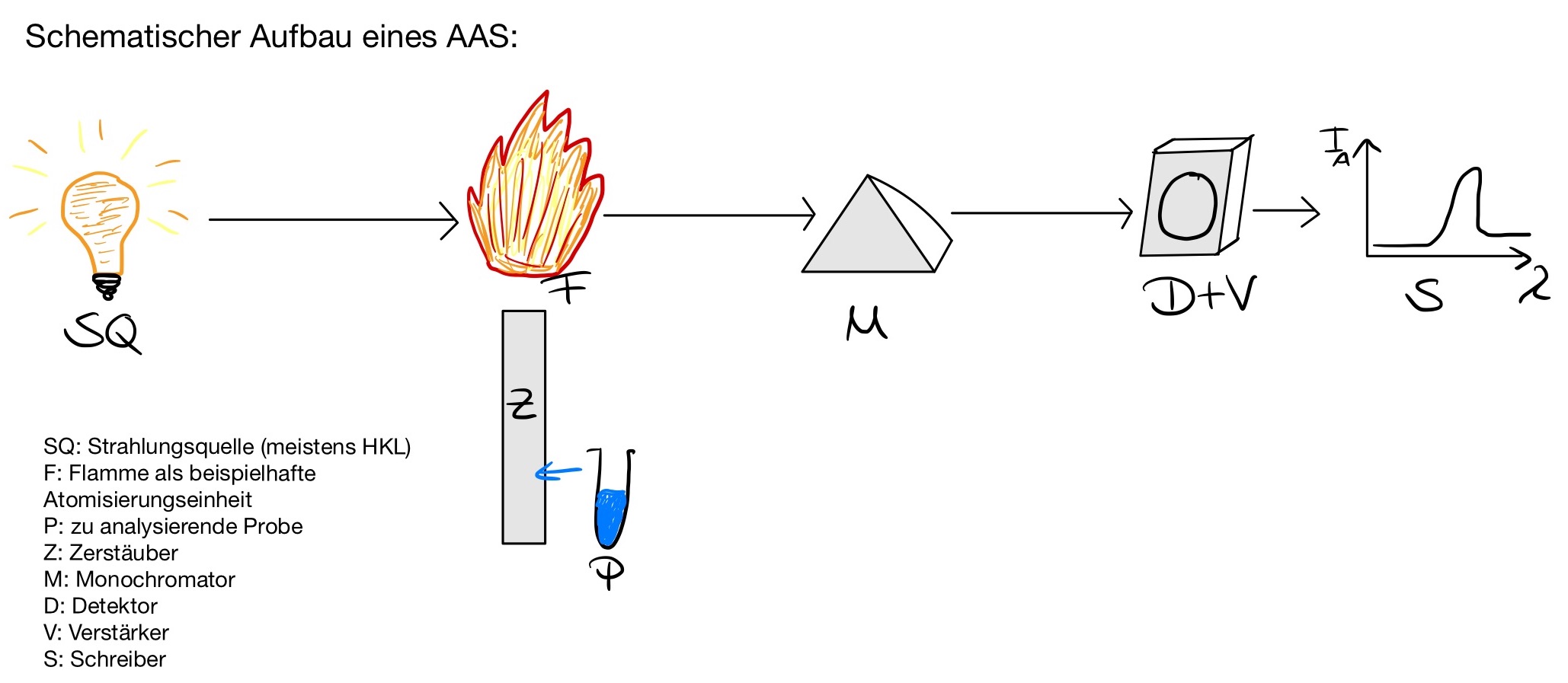
Ein Atomabsorptionsspektrometer, auch AAS-Gerät genannt, setzt sich aus mehreren Komponenten zusammen. Zunächst besteht es aus einer Strahlungsquelle (SQ), normalerweise eine Hohlkathodenlampe, welche die elektromagnetische Strahlung produziert. Daran schließt sich der Atomisator (F) an, in welchen die Probe injiziert und atomisiert wird. Um die zu messende Strahlungslinie zu isolieren, nutzt man einen Monochromator (M), welcher vor oder nach der Atomisierungseinrichtung angeordnet werden kann. Die zur Messung geeignete Resonanzlinie wird schließlich zum Detektor (D) geleitet, welcher die Strahlung misst und als Signal auf dem Anzeigeinstrument (S) abbildet.9
Strahlungsquelle
Die Strahlungsquelle erzeugt genau die Emissionslinien, welche zu den spezifischen Resonanzlinien des zu bestimmenden Elements in der Probe passen. Um für die Messung geeignet zu sein, muss die gewählte Strahlungsquelle bestimmte Anforderungen erfüllen.
Zunächst muss die Messlinie ausreichend von anderen Emissionslinien isoliert sein. Außerdem muss ihre Linienbreite, welche temperaturabhängig ist, deutlich geringer sein als die Linienbreite der Atomabsorptionslinie des zu bestimmenden Elements in der Probe. Darüber hinaus muss die Intensität der Messlinie genügend stark und konstant sein.10
Hohlkathodenlampe

Die meistverwendete Strahlungsquelle ist die Hohlkathodenlampe (HKL). Diese besteht aus einer Wolfram- oder Nickelanode und einer Kathode in Form eines Halbzylinders, welche entweder selbst aus dem zu bestimmenden Element besteht oder von diesem überzogen beziehungsweise mit diesem befüllt ist. Geschützt wird die Kathode mit einem Glaskolben. Anode und Kathode befinden sich in einem Glaszylinder mit UV-durchlässigem Quarzfenster, der bei einem Druck von 1 bis 5 Torr mit Neon oder Argon gefüllt ist.
Die Hohlkathodenlampe erzeugt das Emissionsspektrum des entsprechenden Elements aufgrund einer elektrischen Glimmentladung. Dazu wird eine Spannung von ca. 400 V angelegt. Dies führt zu einer Glimmentladung zwischen den beiden Elektroden, sodass das Füllgas ionisiert wird. Es kommt zu einem Stromfluss (ca. 100 mA) und die Ionen des Edelgases werden zur Kathode beschleunigt. Beim Auftreffen schlagen sie Metallatome aus der Oberfläche der Kathode heraus. Die Metallatome werden dabei teilweise angeregt und emittieren das Licht. Dieses trifft im Atomisator auf die zu analysierende Probe, welche die Strahlung im Bereich der Resonanzlinien absorbiert.
Da die Kathode der HKL aus dem zu analysierendem Element besteht, ist das ausgesendete Lichtbündel geometrisch eng begrenzt und schafft eine hohe Selektivität. Außerdem ermöglicht die HKL eine hohe und konstante Strahlungsintensität. Nachteilig ist, dass sich die Kathode der Lampe durch das Herausschlagen von Atomen verbraucht. Dadurch ist die HKL ungeeignet für leicht flüchtige Substanzen. Außerdem ist für jede Bestimmung eines Elements eine eigene Lampe nötig. Es gibt jedoch auch HKL, deren Kathode mit mehr als einem Metall belegt ist, sodass die Analyse mehrerer Metalle möglich ist.12
Elektrodenlose Entladungslampe
Die Elektrodenlose Entladungslampe (EDL) ist eine Spezialform der HKL und eignet sich nur für die Bestimmung leicht flüchtiger Elemente (As, Se, Sb, Na, K, Pb). Sie enthält keine Elektroden, sondern arbeitet zur Energiebereitstellung mit einem Hochfrequenzfeld oder Mikrowellenfeld. Die EDL bietet eine hohe Strahlungsdichte bei gleichzeitig geringer Linienbreite und im Vergleich zu der HKL ein besseres Signal-Rausch-Verhältnis. Außerdem ist sie langlebiger und stabiler, da keine Elektroden verbraucht werden und dieselbe Lampe für die Vermessung unterschiedlicher Substanzen verwendet werden kann. Die EDL ist aufgrund der höheren Empfindlichkeit und niedrigeren Nachweisgrenzen für die Vermessung bestimmter Elemente, beispielsweise As, Se und Te, besser geeignet als die HKL.13
Xenonkurzbogenlampe
Die Xenonkurzbogenlampe nutzt die Strahlungsemission eines ionisierten Lichtbogens und verfügt über eine hohe Strahlungsdichte über den gesamten relevanten Spektralbereich (Unterschied zur HKL, welche nur den Spektralbereich eines Elements abdeckt).
Sie besteht aus einem dickwandigen Quarzglaskolben, welcher mit Xenon befüllt ist. Darin sind zwei Wolframelektroden eingeschmolzen, zwischen denen ein Abstand von wenigen Millimetern besteht. Da bei Betrieb Temperaturen von 10.000 K (~9723 °C) erreicht werden können, ist die Anode zur Kühlung mit Wasserkanälen durchzogen. Um die Austrittsarbeit der Elektronen und somit die Zündspannung zu reduzieren, enthält die Kathode bis zu 3% 232Thorium. Die Gasfüllung besteht entweder aus reinem Xenon oder ist mit einem geringen Anteil an Quecksilber versetzt.
Die Xenonkurzbogenlampe wird mit kurzen, aufeinander folgenden Hochspannungsimpulsen von 10-60kV gezündet, woraufhin die Gasstrecke zwischen den beiden Elektroden ionisiert wird. Bei ausreichend leistungsstarker Stromversorgung bildet sich ein ionisierter Lichtbogen zwischen den Elektroden. Dieser muss durch einen kontinuierlichen Gleichstrom stabilisiert werden, da der Lichtbogen einen negativen differentiellen Widerstand hat.
Die Lebensdauer der Xenonkurzbogenlampe kann über 1000 Stunden betragen.14
Atomisierungsverfahren
In der pharmazeutischen Analytik werden vier Techniken zur Atomisierung der Probelösung unterschieden. Sehr relevant sind dabei die Flammentechnik und die flammenlose Graphitrohrtechnik. Randtechniken stellen die Hydrid- und ihre Unterform die Kaltdampf-Technik dar.
Ziel dieser Techniken ist die Erzeugung von Atomen in der Gasphase.
Flammen-Technik (F-AAS)
Eine wässrige Untersuchungslösung ist sehr gängig in dieser AAS-Technik, diese wird versprüht und in der Brennerflamme atomisiert. In dieser Flamme verdampft die Probelösung und wird thermisch zersetzt. Je nachdem welches Element bestimmt werden soll, schreibt das Arzneibuch verschiedene Brenngase vor.
In speziellen Zerstäubern wird die zu analysierende Lösung versprüht. Die so entstandenen Nebeltröpfchen werden mit dem Gasstrom in die Brennkammer geleitet. Sollten größere Tröpfchen entstanden sein, so werden diese als Kondensate entfernt. Das Brenngas, z.B. Acetylen, und ein Oxidationsmittel, z.B. Luftsauerstoff, werden mit den Nebeltröpfchen in der Brennkammer vermischt. Dort verdampft das Lösungsmittel Wasser als Gas in der Flamme. Folglich bleiben im Aerosol des Gasstroms feste Teilchen zurück. Unerwünscht wäre hier die Bildung von Oxiden. Durch weitere Flammenhitze werden die Teilchen im Aerosol flüssig bzw. gasförmig, dann zerfallen sie zu Molekülen und schlussendlich zu Atomen. Zusammenfassend ist die Aufgabe der Flammen-Technik, durch die Flammenhitze einen möglichst hohen Anteil des zu bestimmenden Elements in den atomaren Zustand zu bringen. 15 16
Graphitrohr-Technik (GF-AAS)
Eine andere Methode zur elektrothermischen Atomisierung der Probesubstanz ist es, einen Graphitrohrofen einzusetzen. In diesem Ofen halten zwei gekühlte Graphitkontakte, durch welche die Stromzufuhr erfolgt, das Graphitrohr. Durch das Anlegen verschiedener Spannungen und Stromstärken können mit Hilfe von Widerstandsheizungen sehr hohe Temperaturen von bis zu 3000K bzw. 2727°C im Ofen erreicht werden. Das Verbrennen des Graphitrohres wird durch Argon als Schutzgas verhindert. Durch ein Quarzfenster am Graphitrohr wird das, in der Regel von der Hohlkathodenlampe, emittierte Licht eingestrahlt. Mit Hilfe eines Mikropipettiersystems werden sehr geringe Mengen des Analyten in das Rohr injiziert. Über ein Temperaturprogramm wird die Probe zuerst getrocknet, wobei das Lösemittel verdampft. Um störende Begleitsubstanzen thermisch zu entfernen, wird eine weitere Erhitzung vorgenommen, woraufhin die eigentliche Atomisierung erfolgt. Im Anschluss wird das Rohr ausgeheizt, um Restbestandteile der Probe zu entfernen. Zusammenfassend besteht die Graphitrohr-Technik aus den Schritten: 1.Trocknen, 2.Veraschen/Pyrolyse (Zerstörung der Matrixbestandteile, vor allem organischer), 3.Atomisieren und 4.Ausheizen.
Der Vorteil der flammenlosen AAS ist, dass störende Stoffe durch ein Temperaturprogramm entfernt werden können, wodurch die Konzentrierung des atomaren Dampfs zunimmt. Zudem wird eine geringere Probenmenge als bei der Flammen-Technik benötigt und die Nachweisgrenze des Verfahrens ist deutlich verbessert; das erklärt auch den Einsatz in der Spurenanalytik. Durch die Induktionsheizung können sogar schwer flüchtige Metalle in die Gasphase überführt werden. 17 18
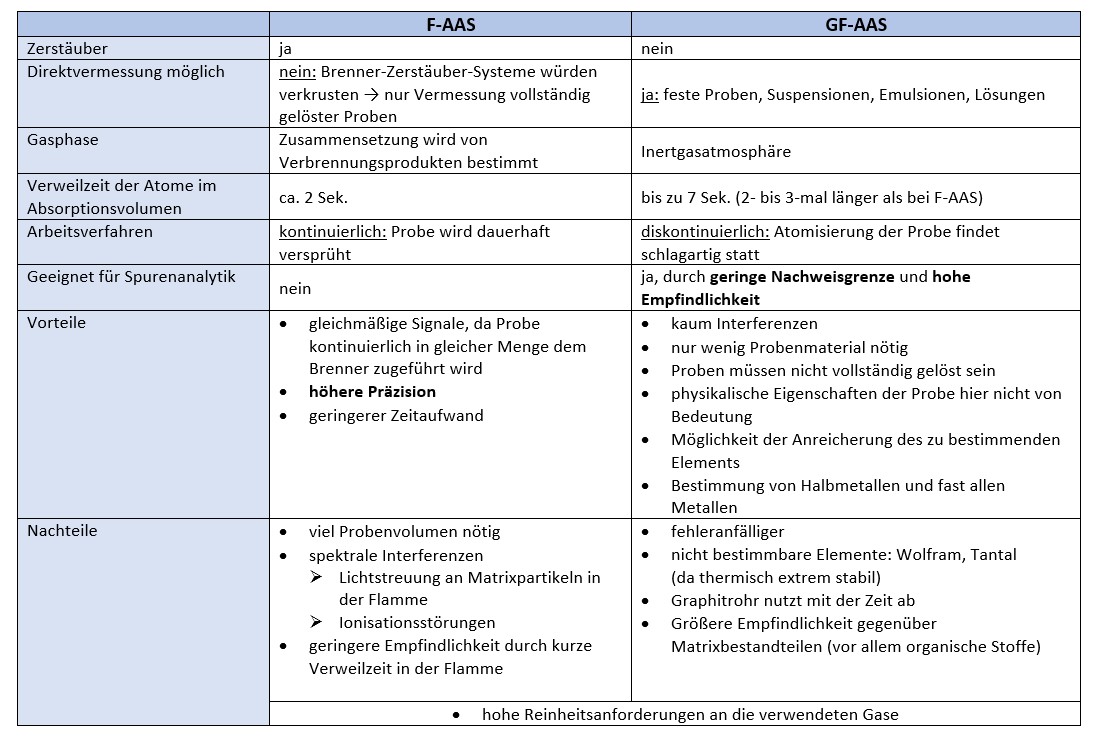
Vergleich Flammen-AAS und Graphitrohr-AAS
Hydrid-Technik
Bei dieser Technik stellt eine auf 850-1000°C beheizte Quarzküvette die Atomisierungseinheit dar. Ausgewählte Elemente lassen sich nach der Reduktion, z.B. mit Natriumborhydrid, als Hydride verflüchtigen und somit von störenden Stoffen befreien. Zu diesen ausgewählten Elementen zählen Arsen, Antimon, Bismut, Selen, Tellur und Zinn. Durch thermische Zersetzung der Probe erfolgt dann die Atomisierung in der Quarzküvette. Das flüchtige Hydrid zerfällt bei den hohen Temperaturen in die Atome des betreffenden Elements und Wasserstoff. 21 22
Kaltdampf-Technik
Hierbei handelt es sich um eine Unterform der Hydrid-Technik, die ausschließlich für die Bestimmung von Quecksilber geeignet ist. Wegen des hohen Dampfdruckes ist Quecksilber das einzige Element, das in metallischer Form bestimmt und mit einem Inertgasstrom ausgetrieben werden kann. Zur Erhöhung der Empfindlichkeit lässt man die Quecksilberverbindung zunächst bei Raumtemperatur auf z.B. Gold als Amalgam niederschlagen (chemische Reduktion); somit wird das elementare Quecksilber gesammelt und angereichert. Durch schnelles Erhitzen wird nun das Quecksilber durch das Trägergas in die Quarzküvette transportiert.
Eine Messung bei Raumtemperatur ist möglich, jedoch erfolgt diese meistens bei ca. 100°C, um Wasserablagerungen in der Küvette zu vermeiden. 23 24 25
Monochromator
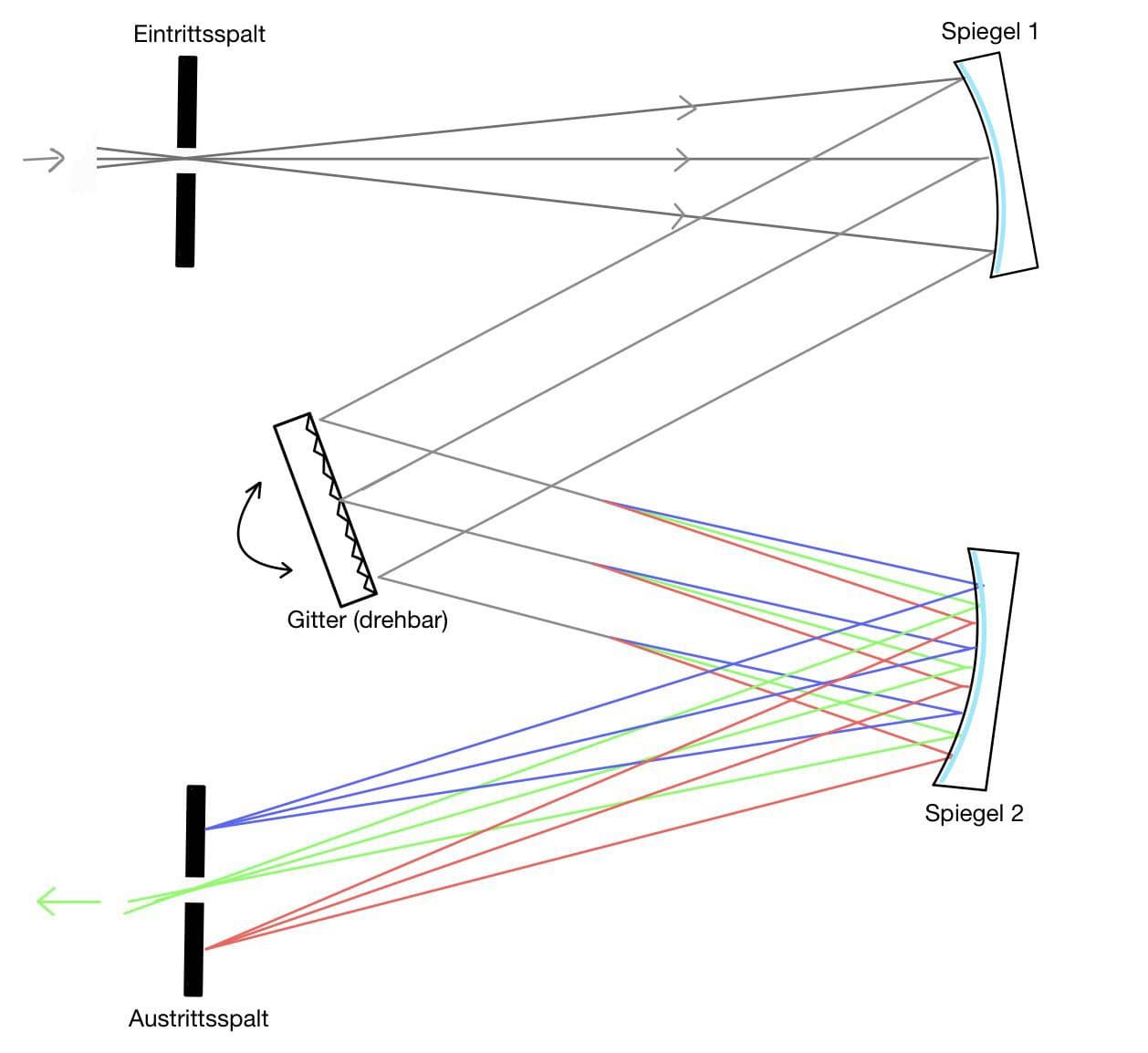
Der Monochromator teilt das Licht aus der Lampe und der leuchtenden Atomisierungseinheit in sein Spektrum auf und isoliert daraus eine bestimmte Wellenlänge. Er besteht in der AAS meist aus einer Anordnung von Gittern und Spiegeln zur Aussonderung einzelner Linien.
Bei der AAS erzeugen die Strahlungsquellen kein kontinuierliches Spektrum, sondern nur die Spektrallinien des enthaltenen Elements (s. z.B. Hohlkathodenlampe). Bei diesen Linienstrahlern als Strahlungsquelle sind keine hochauflösenden Monochromatoren notwendig, da diese nur eine Linie aussondern müssen. Dennoch kann auf einen Monochromator nicht verzichtet werden, da die Strahlungsquelle kein monochromatisches Licht erzeugt, sondern ein Gemisch aller im Linienspektrum des Elements enthaltenen Wellenlängen.
Für die Messung wird eine möglichst intensive Emissionslinie ausgewählt, die aber idealerweise nur von dem zu bestimmenden Element absorbiert wird.
27 28
Detektor
Hinter dem Monochromator wird zur Messung der Abschwächung der Intensität des Primärlichtes ein Detektor eingesetzt. Über diesen Detektor wird das Absorptionssignal nach Verstärkung und Umwandlung mit einem Signalschreiber aufgezeichnet.
Bei den eingesetzten Detektoren handelt es sich meist um Sekundärelektronenvervielfacher (SEV) oder Halbleiterdetektoren. Halbleiterdetektoren zeigen ein besseres Signal/Rausch-Verhältnis, da sie über den vermessenen Spektralbereich (190-900 nm) eine homogenere Quanteneffizienz (Lichtausbeute) haben. Dies wird deutlich durch bessere Nachweisgrenzen.
29 30 31
Interferenzen in der AAS
Spektrale Interferenzen
Es wird in der Regel die strahlungsintensivste Spektrallinie des Analyten für die Messung genutzt. Beträgt ihr Abstand zu der benachbarten Spektrallinie eines anderen Elements weniger als 0,01 nm, kommt es zu einer zusätzlichen Abschwächung der gemessenen Strahlung. Um diese Überlappungen zu vermeiden, kann man entweder die Spaltbreite des Monochromators verringern oder eine andere Spektrallinie des Analyten zur Messung verwenden.
Untergrundabsorption entsteht durch zusätzliche Absorption von Begleitsubstanzen oder Streuung an Verbrennungsprodukten der Matrix wie z.B. Ruß oder Oxide. Sie kann apparativ durch die D2-Untergrundkorrektur (bei F-AAS) oder die Zeeman-Untergrundkorrektur (bei GF-AAS) kompensiert werden.
Deuterium (D2)- Untergrundkompensation
Neben der Hohlkathodenlampe wird zusätzlich eine Deuteriumlampe (Wellenlänge 0,2 nm) angebracht und mit einem Sektorspiegel abwechselnd das Licht des Linienstrahlers und das des Kontinuumstrahlers zur Probe durchgelassen. Breitbandige, unspezifische Lichtverluste führen in beiden Lichtquellen zur gleichen Abschwächung, sodass durch die Deuteriumlampe nur die Untergrundabsorption und durch die Hohlkathodenlampe die Gesamtabsorption gemessen wird. Die Differenz der beiden ergibt die Atomabsorption.
Zeeman-Untergrundkompensation
Hier befindet sich das Graphitrohr (s. o. GF-AAS) in einem starken Magnetfeld, wodurch die Energieniveaus einzelner Elektronenzustände verschoben werden. So werden die Spektrallinien aufgespalten.
Man unterscheidet zwischen dem transversalen Zeemanneffekt, der bei Beobachtung bzw. Strahlungsrichtung senkrecht zum Magnetfeld zu zwei verschobenen Komponenten ( Δm = ± 1) und einer linear polarisierten Komponente (Δm = 0) der Emission führt, und dem longitudinalen Zeemanneffekt bei Beobachtung bzw. Strahlungsrichtung parallel zum Magnetfeld, bei dem zwei verschobene, entgegengesetzt zirkulär polarisierte Linien auftreten ( Δm +1/-1). Diese stellen die elektrische Dipolschwingung dar.
Die verschobenen Linien der beiden Effekte liegen nicht mehr in der Emissionslinie der Lampe und die linear polarisierte Komponente des transversalen Effekts wird mit einem Polarisator abgefangen, somit wird bei eingeschaltetem Magneten nur die Untergrundabsorbtion gemessen. Das Magnetfeld wird abwechselnd ein- und ausgeschaltet (gepulstes Magnetfeld), da bei ausgeschaltetem Magneten nicht nur die Untergrundabsorption, sondern auch die spezifische Absorption gemessen wird. Zieht man nun den Messwert bei eingeschaltetem Magneten vom Messwert bei ausgeschaltetem Magneten ab, erhält man die spezifische Absorption.
Definition Δm: positiv=energieerhöhend, negativ=energieerniedrigend. Der Buchstabe m steht im Orbitalmodell für die Magnetquantenzahl, die das Verhalten eines Elektrons in einem von außen angelegten Magnetfeld beschreibt.
Nicht-spektrale Interferenzen
Transportinterferenzen kommen durch Störungen bei der Zerstäubung von Prüf- und Kalibrierlösungen zustande, wenn diese unterschiedliche physikalische Eigenschaften (Viskosität, Dichte, Oberflächenspannung) besitzen und somit durch unterschiedliche Ansaugraten die Wirksamkeit des Zerstäubers beeinflussen. Aus den gleichen Gründen kann außerdem durch unterschiedliche Tröpfchengrößen der Probeneintrag in die Flamme variieren.
Ausgeglichen werden kann dies durch Angleichen von Kalibrier- und Prüflösung oder durch die Standardadditionsmethode.
Verdampfungsinterferenzen kommen vor, wenn bei Nebenreaktionen des Analyten mit Proben- und Flammenbestandteilen sowie Lösungsmitteln schwer atomisierbare und thermisch stabile Komplexe (Carbide, Oxide, Phosphate) gebildet werden. Dadurch stehen weniger Atome für die Messung zur Verfügung.
Durch höhere Flammentemperaturen oder Abfangreagenzien wie EDTA oder Lanthan-Verbindungen kann dieses Problem gelöst werden.
Ionisationsstörungen treten vor allem bei leicht ionisierbaren Erdalkali- und Alkalimetallen auf, da hier das Gleichgewicht zwischen Atomen und Ionen bei zu hohen Temperaturen auf Seite der Metallionen liegt. Diese haben allerdings ein anderes Absorptionsspektrum als die zugehörigen Metallatome, wodurch das Messergebnis niedriger ausfällt.
Durch Senken der Temperatur oder Zugabe leicht ionisierbarer Verbindungen die als Ionisationspuffer dienen, können Ionisationsstörungen vermieden werden. 32 33
Einzelnachweise
1 https://www.science.org.au/fellowship/fellows/biographical-memoirs/alan-walsh-1916-1998 abgerufen: 17.Mai 2021, 18:35 Uhr ⇑
2 Dominik/Steinhilber/Wurglics; Instrumentelle Analytik kompakt; 3. Auflage 2013 ⇑
3 Dominik/Steinhilber/Wurglics; Instrumentelle Analytik kompakt; 3. Auflage 2013 ⇑
4 Dominik/Steinhilber/Wurglics; Instrumentelle Analytik kompakt; 3. Auflage 2013 ⇑
5 Rücker/Neugebauer/Williams; Instrumentelle pharmazeutische Analytik; 4. Auflage, 2008 ⇑
6 Dominik/Steinhilber/Wurglics; Instrumentelle Analytik kompakt; 3. Auflage 2013 ⇑
7 Rücker/Neugebauer/Williams; Instrumentelle pharmazeutische Analytik; 4. Auflage, 2008 ⇑
8 Angelehnt an: Schwedt, Georg; Analytische Chemie: Grundlagen, Methoden und Praxis; 2. Auflage, 2008; gezeichnet von Maja Dominique Voßgröne ⇑
9 Rücker/Neugebauer/Williams; Instrumentelle pharmazeutische Analytik; 4. Auflage, 2008 ⇑
10 Rücker/Neugebauer/Williams; Instrumentelle pharmazeutische Analytik; 4. Auflage, 2008 ⇑
11 Schwedt, Georg; Analytische Chemie: Grundlagen, Methoden und Praxis; 2. Auflage, 2008 ⇑
12 Rücker/Neugebauer/Williams; Instrumentelle pharmazeutische Analytik; 4. Auflage, 2008 ⇑
13 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/16/anac/aasvirtmess.vlu/Page/vsc/de/ch/16/anac/aas5_st_linie.vscml.html abgerufen am 26. Mai, 17:30 Uhr ⇑
14 https://de.wikipedia.org/wiki/Xenon-Gasentladungslampe abgerufen: 26. Mai 2021, 18:00 Uhr ⇑
15 Schwedt, Georg; Analytische Chemie: Grundlagen, Methoden und Praxis; 2. Auflage, 2008 ⇑
16 Ehlers, Eberhard; Analytik II: Kurzlehrbuch Quantitative und Instrumentelle Pharmazeutische Analytik; 12. Auflage, 2016 ⇑
17 Schwedt, Georg; Analytische Chemie: Grundlagen, Methoden und Praxis; 2. Auflage, 2008 ⇑
18 Ehlers, Eberhard; Analytik II: Kurzlehrbuch Quantitative und Instrumentelle Pharmazeutische Analytik; 12. Auflage, 2016 ⇑
19 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/16/anac/aasvirtmess.vlu.html abgerufen: 04. Juni 2021, 17:00 Uhr ⇑
20 Skript; Atomspektroskopische Analyseverfahren; Christin Scheller; SoSe 2020 ⇑
21 Schwedt, Georg; Analytische Chemie: Grundlagen, Methoden und Praxis; 2. Auflage, 2008 ⇑
22 Ehlers, Eberhard; Analytik II: Kurzlehrbuch Quantitative und Instrumentelle Pharmazeutische Analytik; 12. Auflage, 2016 ⇑
23 Schwedt, Georg; Analytische Chemie: Grundlagen, Methoden und Praxis; 2. Auflage, 2008 ⇑
24 Ehlers, Eberhard; Analytik II: Kurzlehrbuch Quantitative und Instrumentelle Pharmazeutische Analytik; 12. Auflage, 2016 ⇑
25 Rücker/Neugebauer/Williams; Instrumentelle pharmazeutische Analytik; 4. Auflage, 2008 ⇑
26 Angelehnt an: https://mindsculpt.me/czerny-turner-monochromator-73/ abgerufen: 17. Juni 2021, 17:00 Uhr; gezeichnet von Meret Krull ⇑
27 Dominik/Steinhilber; Instrumentelle Analytik: Kurzlehrbuch und kommentierte Originalfragen für Pharmazeuten; 2. Auflage, 2002 ⇑
28 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/16/anac/aasvirtmess.vlu/Page/vsc/de/ch/16/anac/aas5_mon.vscml.html abgerufen: 26. Mai 2021, 21:00 Uhr ⇑
29 https://www-app.uni-regensburg.de/Fakultaeten/CHP/Analytische_Chemie/web/dateien/duerkop/AAS.pdf abgerufen: 31. Mai 2021, 10:30 Uhr ⇑
30 https://de.wikipedia.org/wiki/Atomabsorptionsspektrometrie#Detektor abgerufen: 31. Mai 2021, 10:30 Uhr ⇑
31 Schwedt, Georg; Analytische Chemie: Grundlagen, Methoden und Praxis; 2. Auflage, 2008 ⇑
32 https://de.wikipedia.org/wiki/Zeeman-Effekt#Spektroskopie abgerufen: 25.Mai 2021, 20:20 Uhr ⇑
33 http://dodo.fb06.fh-muenchen.de/maier/analytik/Blaetter/N091_Atomabsorptionsspektrometrie1_a_BAneu.pdf abgerufen: 25.Mai 2021, 20:00 Uhr ⇑
Monographiebeispiel: Omeprazol-Magnesium (Prüfung auf Reinheit, Gehalt an Magnesium)
Stoffcharakterisierung

Die Substanz liegt als weißes bis fast weißes, hygroskopisches Pulver vor und ist in Wasser nur sehr schwer löslich, wenig löslich in Methanol und praktisch unlöslich in Heptan.2
„Omeprazol-Magnesium besitzt ein chirales Zentrum in der Sulfinylgruppe, demgemäß gibt es zwei Enantiomere. Das Ph. Eur. beschreibt das Racemat" (Ph. Eur. 6.7/2374)
Wirkung
Omeprazol-Magnesium ist ein Benzimidazolderivat und gehört zu den Protonenpumpeninhibitoren (H+/K+-ATPase-Blocker, kurz: PPI). Es wirkt sich auf die Säureproduktion im Magen aus, indem es die H+/K+-ATPasen der Belegzellen der Magenschleimhaut irreversibel inaktiviert. Durch Hemmung des terminalen Schrittes der Salzsäureproduktion der Belegzellen wird die intragastrische Azidität reduziert und der pH-Wert erhöht. Omeprazol-Magnesium wird zügig im Dünndarm resorbiert, sodass bereits nach 0,5 bis 3,5 Stunden eine maximale Plasmakonzentration erreicht werden kann.
Der Arzneistoff reichert sich in Form einer schwachen Base als Prodrug im sauren Bereich der Belegzellen an und entfaltet seine Wirkung als Enzymhemmer erst durch Protonierung. Im sauren Milieu (pH-Wert < 4) wandelt sich das Omeprazol-Magnesium zum wirksamen Sulfenamid um. Dieser Metabolit reagiert dann mit der Protonen-Kalium-ATPase durch Ausbildung einer Disulfidbrücke, wodurch das Enzym irreversibel blockiert wird. Eine Regeneration des Enzyms findet nur durch eine De-novo-Synthese statt.
Die Hemmung der Protonen-Kalium-ATPase der Belegzellen und der damit verbundenen Erhöhung des pH-Wertes verhindert gleichzeitig einen Überschuss an Omeprazol-Magnesium in der Zelle, da sich durch den erhöhten pH-Wert weniger Omeprazol-Magnesium in den wirksamen Metaboliten umwandelt. Die Biotransformation ist somit über eine Art Feedback-Mechanismus geregelt. 3
Anwendung
Omeprazol-Magnesium wird bei folgenden Beschwerden angewendet:
- Sodbrennen
- Reizmagen
- Gastritis
- bei Einnahmen hoher Dosen an Schmerzmitteln wie Ibuprofen als Schutz, da diese die Magensäuresynthese erhöhen
- Infektionen mit Helicobacter pylori, zusammen mit Antibiotikakombination (Amoxicillin/Clarithromycin) 4
Durchführung der Monographie
Laut Europäischem Arzneibuch wird für die Überprüfung auf Reinheit nach Methode I verfahren. Das heißt die Konzentrationsbestimmung erfolgt mithilfe der Atomabsorptionsspektroskopie durch eine direkte Kalibrierung mit drei Referenzlösungen, deren Gehalt dem 0,7- bis 1,3-fachen Gehalt der Untersuchungslösung entsprechen. 5
Zur Herstellung der Untersuchungslösung werden zunächst 0,250 g Substanz langsam mit 20,0 mL Salzsäure (103 g/L) gelöst. Anschließend wird diese Lösung mit Wasser zu 100,0 mL verdünnt. Davon werden 10,0 mL mit Wasser zu 200,0 mL erneut verdünnt. Aus dieser zweiten Verdünnung werden 10,0 mL entnommen und mit 4 mL Lanthan(III)-chlorid-Lösung versetzt und abschließend zu 100,0 mL verdünnt.6
Für die Herstellung der Referenzlösungen macht das Europäische Arzneibuch folgende Angaben:
„Referenzlösungen werden aus Magnesium-Lösung (1000 ppm Mg) R durch Verdünnen mit einer Mischung von 1 mL einer Lösung von Salzsäure R (103 g/L) und 1000,0 mL Wasser R hergestellt." (Ph. Eur. 10.0/2374)
Zur Bestimmung des Gehaltes an Magnesium wird die Abschwächung der Lichtintensität in einen proportionalen Zusammenhang zur Konzentration der absorbierenden Magnesiumatome gesetzt. Damit kann über die Anwendung des Lambert-Beer'schen Gesetzes und der vermessenen Absorption der Gehalt an Magnesium errechnet werden.
Auswertung/ Interpretation/ Bedeutung
Die quantitative Auswertung des Verfahrens beruht auf der Anwendung des Lambert-Beer'schen Gesetzes. Vermessen wird die Strahlungsenergie, die von den Atomen des zu bestimmenden Elementes absorbiert wird. Daraus resultiert eine Lichtabschwächung, die zu der Konzentration der absorbierenden Metallatome in der Probe direkt proportional ist.
Das Absorptionsmaximum der Prüflösung für Magnesium muss bei 285,2 nm liegen. Damit die Substanz den Anforderungen des Arzneibuchs entspricht, muss sich aus der Vermessung ein Gehalt von 3,30 bis 3,55 % an Magnesium in wasserfreier Substanz ergeben. Bei Werten außerhalb dieser Gehaltsspanne für Magnesium sind die Anforderungen an die Prüfsubstanz nach Europäischem Arzneibuch nicht mehr erfüllt. Der theoretische Wert für den Gehalt an Magnesium liegt bei 3,41 %.7
Eignung der analytischen Methode
Die Atomabsorptionsspektroskopie eignet sich zur Ermittlung der Reinheit und der Bestimmung an Magnesium, da deren Nutzung sowohl zur quantitativen als auch zur qualitativen Analyse geeignet ist. Die AAS wird deshalb oft in der Pharmazie zur Untersuchung von organometallischen Verbindungen eingesetzt. Darüber hinaus wird je nach instrumentellem Aufbau eines AAS-Gerätes eine hohe Selektivität erreicht. Dies gilt vor allem für Hohlkathodenlampen als Strahlungsquelle, da die Kathode aus dem zu analysierendem Element besteht. Zusätzlich sind mit der AAS sehr geringe Nachweisgrenzen (GF-AAS) und hohe Empfindlichkeiten zu erreichen, was sehr gute Voraussetzungen sowohl zur Reinheitsbestimmung als auch zur Quantifizierung darstellt. Durch die einfache Probenvorbereitung lassen sich schnell und präzise (bedingt durch den Geräteaufbau) Ergebnisse mittels AAS erzielen. 8
Einzelnachweise
1 Ph. Eur 10.0/2374 ⇑
2 Ph. Eur. 10.0/2374 ⇑
3 Ph. Eur. 7.7/0942 ⇑
4 Ph. Eur. 6.7/2374 ⇑
5 Eberhard, Ehlers; Analytik II, Kurzlehrbuch Quantitative und Instrumentelle Pharmazeutische Analytik; 12. Auflage 2016 ⇑
6 Ph. Eur. 10.0/2374 ⇑
7 Ph. Eur. 6.7/2374 ⇑
8 Skript; Atomspektrometrische Analyseverfahren; Christin Scheller; SoSe 2020 ⇑
TU-Braunschweig Institut für Medizinische und Pharmazeutische Chemie Seminar: Instrumentelle Analytik Kontakt: tubs@t-kellner.de
