Demo Pol
Titelblatt
Expertenbericht zum Praktikumsversuch
Demo Pol
WiSe 2021/2022
Abgabedatum
16.12.2021
Expertengruppe 15
Iman Bischo
Mahdia Djawadi
Annabell Steinke
Simultanbestimmung Cd, Cu und Zn (Pol.)
Inhaltsverzeichnis
Einleitung
Die Polarographie ist ein voltametrisches Verfahren, bei dem der Stromfluss (I) in Abhängigkeit von einer angelegten Spannung (U) gemessen wird.1 Dabei kommt es zur Polarisation an einer Quecksilber-Tropfelektrode, die graphisch detektiert wird.2 Pharmazeutisch findet diese Methode Einsatz in der qualitativen und quantitativen Analyse von anorganischen und organischen Substanzen mit reduzierenden funktionellen Gruppen. Der Vorteil der Polarographie ist die niedrige Nachweisgrenze und der damit verbundene geringere Substanzbedarf. Die Methode war in den 1980er Jahren vor allem in der Spurenanalytik von Bedeutung, heute werden wegen der gesundheitsschädlichen Wirkung von Quecksilber hauptsächlich spektroskopische und chromatographische Analyseverfahren eingesetzt.3 4
Die wichtigsten polarographischen Verfahren sind:
- Gleichstrompolarographie: Es wird eine gleichmäßig abnehmende beziehungsweise zunehmende Spannung angelegt.
- Wechselstrompolarographie: Es wird ein Wechselstrom angelegt, wodurch Kapazitätsströme eliminiert werden und somit die Nachweisgrenze verbessert wird.
- Pulspolarographie: Es wird bei jedem gebildeten Quecksilbertropfen ein Spannungspuls abgegeben, dies führt auch zu einer verbesserten Nachweisgrenze.
- Normalpulspolarographie: Der Strom wird nur nach dem Spannungspuls gemessen.
- Differentialpulspolarographie: Der Strom wird vor und nach dem Spannungspuls gemessen.5
Grundlagen (physikalische/chemische)
Bei der Polarographie wird eine Spannung angelegt. In der Lösung befinden sich die Analyten. Wenn die angelegte Spannung das Redoxpotential des Analyten erreicht kommt es zum Stromfluss. Genauer gesagt fließt ein Faraday’scher Strom.6 Während der Polarographie liegt jedoch auch noch ein kapazitiver Strom (=ein Ladungsstrom) vor. Er kommt dadurch zustande, dass sich Elektronen in der Arbeitselektrode befinden und die Kationen in der Elektrolytlösung dadurch zur Elektrode fließen und die Anionen von der Elektrode weg. Der kapazitive Strom soll klein gehalten werden, da er die Nachweisgrenze verschlechtert.7 Das Redoxpotential kann man über die Nernst’sche Gleichung berechnen.8
⚠ $$E = E_0 + \frac{0,059}{z} \cdot ln \frac{c(ox)}{c(red)}⚠ $$
- E = Redoxpotential [V]
- E0 = Normalpotential [V]
- z = Anzahl Elektronen
- c (Ox) = Konzentration Oxidationsmittel [mol/l]
- c (Red) = Konzentration Reduktionsmittel [mol/l]
Durch den Stromfluss kommt es zu einem Elektronenaustausch zwischen Analyten und Elektrode.9 An der Arbeitselektrode findet eine Reduktion der Analyten (Oxidationsmittel) aus der Lösung statt: ⚠ $$Ox. + e^{-} ⇌ Red.⚠ $$
Es passieren somit im Praktikum folgende Reaktionen:
⚠ $$Cu^{2+} + 2e^{-} ⇌ Cu⚠ $$
⚠ $$Cd^{2+} + 2e^{-} ⇌ Cd⚠ $$
⚠ $$Zn^{2+} + 2e^{-} ⇌ Zn⚠ $$
An einer Hilfselektrode findet die Oxidation von Quecksilber statt: 10
⚠ $$2Hg ⇌ Hg_2 {^{2+}} + 4e^{-}⚠ $$
Durch die Veränderung des Verhälnis von c(ox)/c(red) entsteht eine Diffusionsschicht zwischen dem Analyten und der Elektrode. Die Diffusionsschicht und der Stromfluss ergeben zusammen einen Diffusionsstrom. Anhand der Ilkovic Gleichung ist zu erkennen, dass der Diffusionsgrenzstrom (also die Höhe der polarographischen Stufe) proportional zur Konzentration des Analyten in der Lösung ist. Somit kann über den Diffusionsgrenzstrom quantitativ der Gehalt bestimmt werden. 11
⚠ $$I_ {d.grenz} = 607 \cdot z \cdot D^{1/2} \cdot m^{2/3} \cdot t^{1/6} \cdot c⚠ $$
- I d.grenz = Diffusionsgrenzstrom [A]
- z = Anzahl Elektronen
- D = Diffusionskoeffizient [cm2 · s-1]
- m = Ausflussgeschwindigkeit des Quecksilbers [g · s-1]
- t = Lebensdauer Quecksilbertropfen [s]
- c = Konzentration des Analyten [mol · cm-3]
In der Praxis lässt sich der Diffusionsgrenzstrom schwer über die Ilkovic-Gleichung berechnen, da der Diffusionskoeffizient schwer zu bestimmen ist.
Aus diesem Grund erfolgt eine graphische Auswertung.
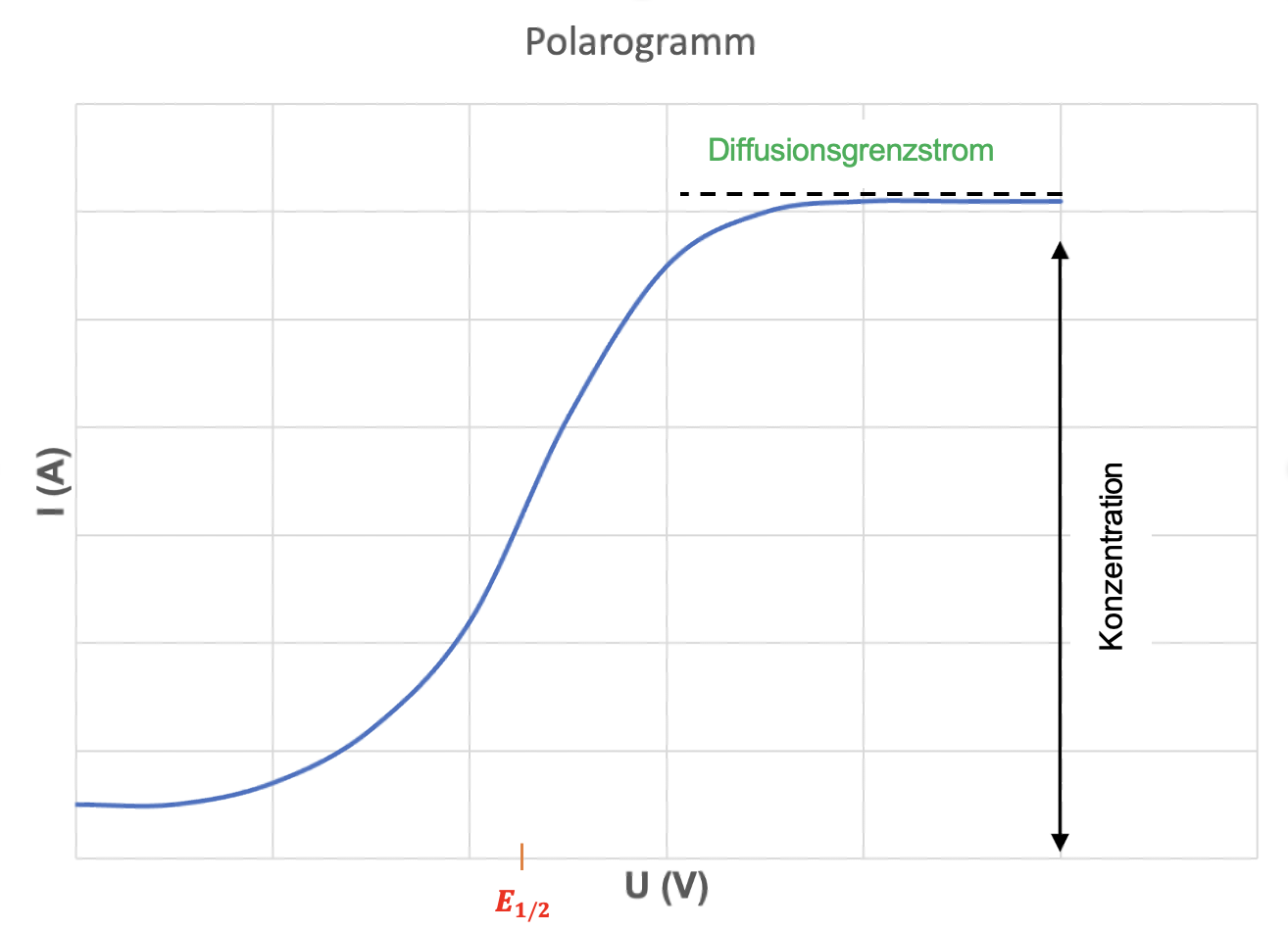
Folgendes ist dabei relevant: bei der Gleistrompolarographie sehen die Polarogramme stufenförmig aus (siehe Abbildung 1).
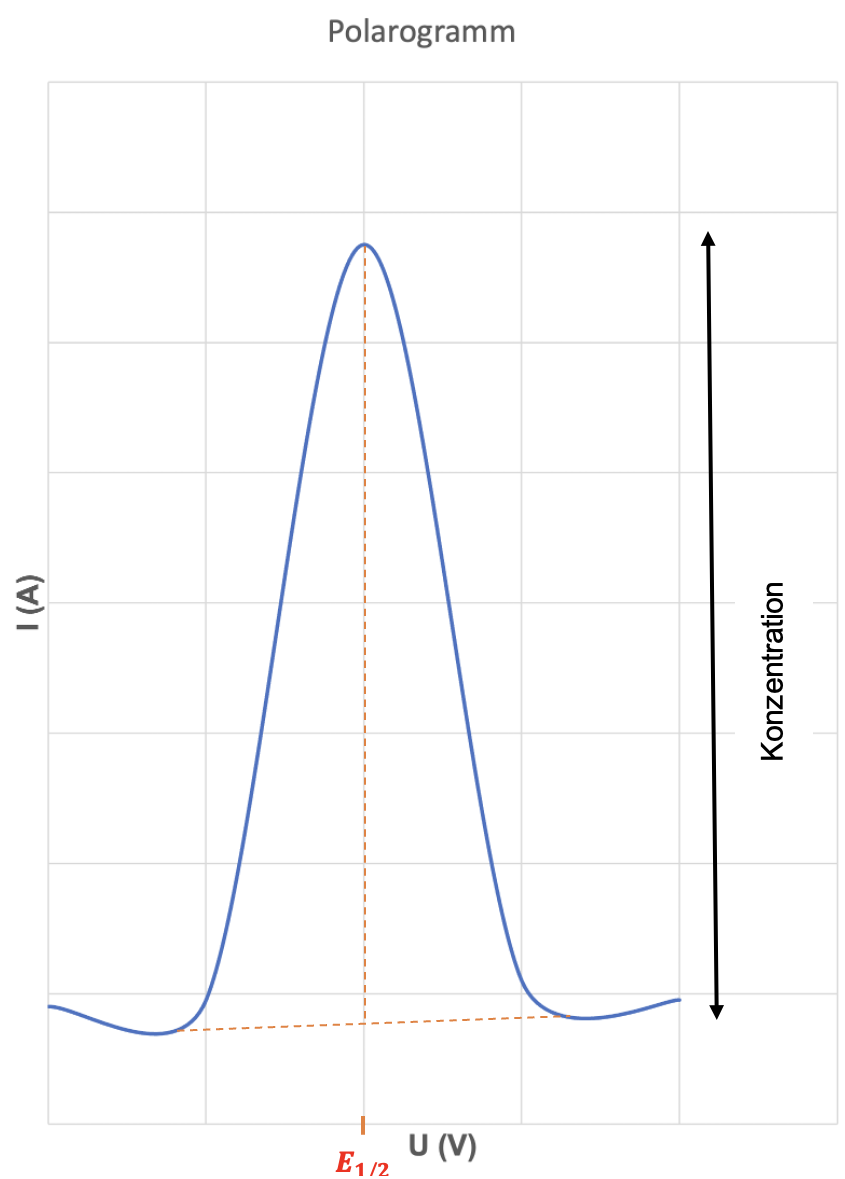
Bei der Wechselstrompolarographie und der Differentialpulspolarographie hat man peakförmige Polarogramme (siehe Abbildung 2).
Bei den stufenförmigen Polarogrammen erhält man die Konzentration über die Stufenhöhe und bei den peakförmigen Polarogrammen erhält man die Konzentration über die Peakhöhe. Anhand des Halbstufenpotentials kann man den Analyten identifizieren.
Wie man an der Abbildung 1 auch sehen kann, ist bei den stufenförmigen Polarogrammen das Halbstufenpotential die Spannung, die man auf halber Stufenhöhe abliest. Hingegen ist bei den peakförmigen Polarogrammen das Halbstufenpotential die Spannung, die man von der Peakspitze hinunter gefällten Lot abliest.12
Versuchsbeschreibung
Die polarographische Bestimmung wird als Demonstrationsversuch durchgeführt. Ziel des Versuches ist es, den Gehalt und das Halbstufenpotential von den simultan vorliegenden Kationen Cd 2+ , Cu 2+ , Zn 2+ zu bestimmen. Dazu werden drei Polarogramme ausgewertet. Ein Polarogramm der Standardlösung und zwei Polarogramme mit unterschiedlichen Leitelektrolyten. Vorerst wird ermittelt, welche Peakspitze zu welchem Kation gehören. Die Reihenfolge der auftretenden Peaks ist durch ihr Redoxpotential bestimmt. Als erstes tritt das edelste Element Cu 2+ (+0,34V), danach Cd 2+ (-0,40V) und zum Schluss Zn 2+ (-0,76V) auf.15 Um die Konzentration zu bestimmen, wird die Höhe der Peaks von einer Grundlinie bestimmt und über die Stromstärke die Konzentration ausgerechnet. Zur Bestimmung des Halbstufenpotentials, das bestimmt wird, um einen Stoff zu identifizieren, wird ein Lot von der Peakspitze zur x-Achse gefällt. Vom Spannungswert des Lots wird der Abstand bis zu 0 V ausgemessen und anschließend das Halbstufenpotential ausgerechnet.
Instrumenteller Aufbau
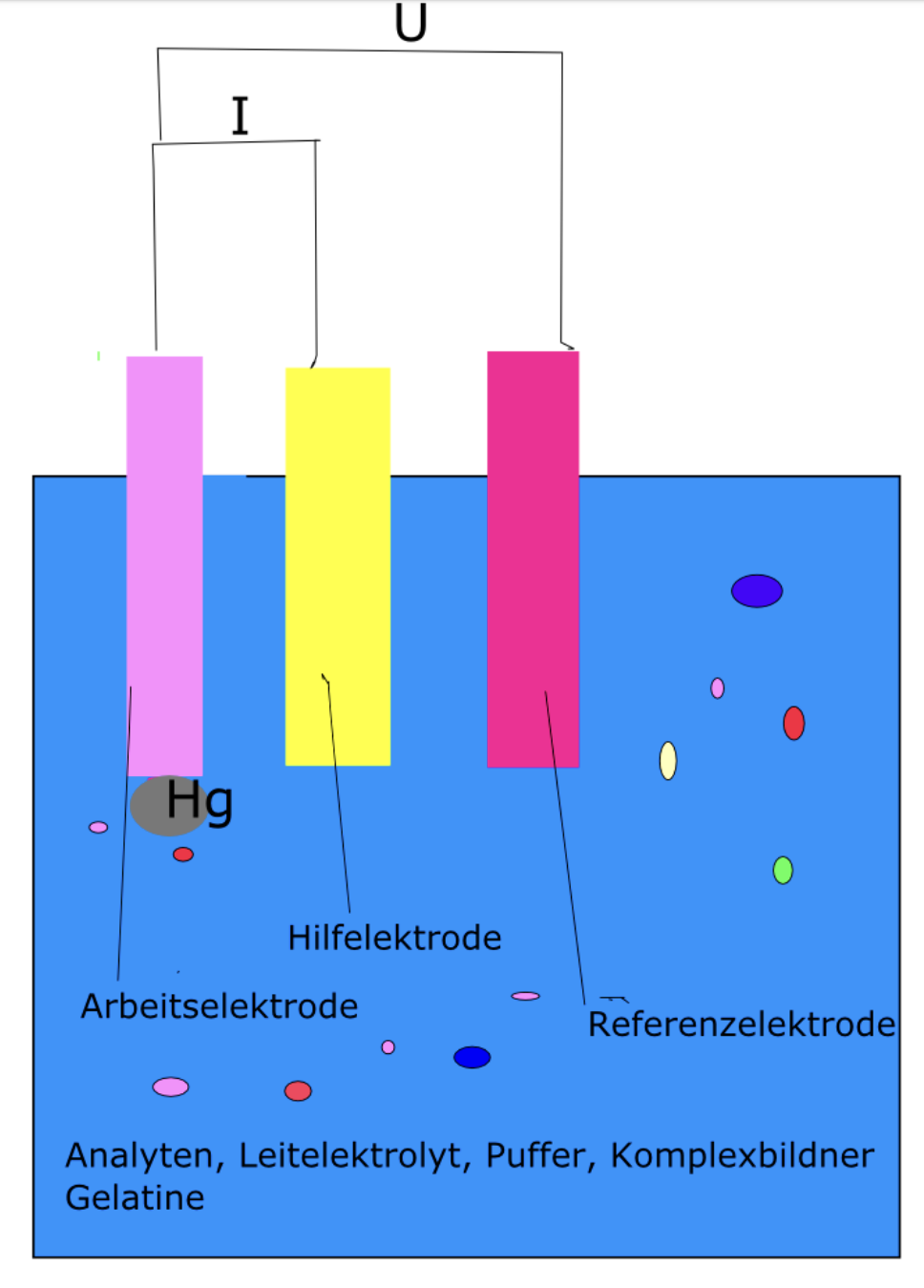

Anhand den Abbildungen 3 und 4 kann man sehen, dass die Messzelle aus drei Elektroden besteht.
- Arbeitselektrode: Die Arbeitselektrode ist eine Quecksilbertropfelektrode. Die Elektrode enthält eine Kapillare, die mit Quecksilber gefüllt ist. Aus der Kapillare tropft ein Tropfen Quecksilber, der in bestimmten Zeiten mit einem Hammer abgeschlagen wird.
- Referenzelektrode: Dabei handelt es sich um eine Silber/ Silberchlorid Elektrode.
- Gegenelektrode/ Hilfselektrode: Dabei handelt es sich um eine Platin- oder Graphitelektrode. 18
Zwischen der Arbeitselektrode und der Hilfslelektrode wird der Strom gemessen und zwischen der Arbeitselektrode und der Referenzelektrode die Spannung.19 Die drei Elektroden tauchen in eine Lösung, die den Analyten, Leitelektrolyt, Puffer, Komplexbildner und Gelatine enthält.
Durchführung
Als Erstes wird die Analysenlösung mit destilliertem Wasser zu 100,0 ml aufgefüllt. Danach werden 25 ml Analysenlösung in einen 50,0 ml Messkolben pipettiert. In den Messkolben werden zwei Tropfen Gelatinelösung hinzugegeben und mit dem Leitelektrolyt Natriumacetat / Essigsäure-Pufferlösung (pH 4,6) zu 50,0ml aufgefüllt. Etwa 20 ml dieser Lösung werden in einen Polarographiebecher gegeben und mit N2 begast. Folgende Daten werden am Polarographen vorprogrammiert:
| Electrode | SMDE | Drop size: 3 | Stirrer speed (rpm): 2000 |
| Initial purge time (s) | 300 | Start potential (V): 0,2 | Endpotential (V): -1,3 |
| Equilibration time | 5 | Pulse amplitude (V): 0,30 | Pulse time (s9). 0,040 |
| Cell off after measurement | Yes | Voltage Stepp (V): 0,005 |
Im Anschluss erfolgt eine Einpunktkalibrierung. Es werden jeweils 5,0 ml der Standardlösungen Cd (250 ppm), Cu (250 ppm) und Zn (250 ppm), 10 ml Wasser und zwei Tropfen Gelatinelösung in einem 50,0 ml Messkolben hineingeführt. Der Messkolben wird mit Natriumacetat/Essigsäure-Pufferlösung (pH= 4,6) zu 50,0 ml aufgefüllt. Danach werden die Polarogramme von der Kalibierlösung und der Analysenlösung aufgenommen. Als Nächstes erfolgt eine weitere Aufnahme eines Polarogramms mit einem anderen Leitelektrolyten. Dafür werden 25 ml Analysenlösung in einen 50,0ml Messkolben pipettiert, mit zwei Tropfen Gelatinelösung versetzt und mit einer NH3/NH4CI-Lösung (1 mol/L) zu 50,0 mL aufgefüllt. Etwa 20 ml der Lösung werden in einen Polarographiebecher gegeben, mit Stickstoff begast und dann das Polarogramm aufgenommen.
Anschließend erfolgt die Entsorgung der Lösungen im Quecksilberabfall. 20
Hintergrundinformationen zur Durchführung
Der Analytlösung ist ein Leitelektrolyt zuzusetzen. Dabei handelt es sich um ein Salz wie z.B. KCl, NaClO4, NaSO4. Dieser Zusatz ist wichtig, um die Migration zu verhindern (Stofftransport geladener Teilchen im elektrischen Feld). Die Analyten sollen nur durch Diffusion zur Elektrode gelangen. Eine Konvektion (Stofftransport durch Durchmischung) muss auch verhindert werden, indem das Rühren der Lösung vermieden wird. Ein Puffer wird hinzugegeben, da Redoxpotentiale pH-abhängig sind. Der Komplexbildner kann Halbstufenpotentiale so verschieben, dass die Analyten sich im Messbereich befinden. Die wenigen Tropfen zugegebene Gelatinelösung verhindert störende Maxima im Bereich der polarographischen Stufen und wird auch als Maximumdämpfer bezeichnet.21 Vor Beginn der Messung wird die Analytlösung noch mit Stickstoff begast, um O2 auszuspülen, da Sauerstoff zwei polarographische Stufen, die sich mit den Stufen der Analyten überlappen, verursachen könnte. Die erste polarographische Stufe befindet sich bei -0,2V und es wird Sauerstoff zu Wasserstoffperoxid reduziert: O2 + 2 H++ 2e- ⇌ H2O2. Die zweite polarographische Stufe befindet sich bei -0,9 V und es wird Wasserstoffperoxid reduziert: H2O2 + 2 H+ + 2e- -> 2 H2O. 22Außerdem wird bei der Tropf-Elektrode die Oberfläche der Elektrode durch die tropfenweise Zugabe von Quecksilber ständig erneuert und reproduzierbar gemacht, um die Wanderung von Teilchen zu verbessern und vor Vergiftungen zu schützen .2324
Gefahr Quecksilber
Auswertung
Es wurden im Versuch drei Polarogramme erhalten:
- Polarogramm zu den Standardlösungen
- Polarogramm Probe; Leitelektrolyt: Na-acetat/Essigsäure (pH = 4,6)
- Polarogramm Probe; Leitelektrolyt: Ammoniak/Ammoniumchlorid (1 mol/L)
Zum Verständnis ist es wichtig zu wissen:
- die erhaltenen Polarogramme sehen peakförmig aus, da eine Differentialpulspolarographie durchgeführt wurde
- durch die unterschiedlichen Leitelektrolyte wird das Halbstufenpotential verschoben
Im ersten Schritt der Auswertung wird die Massenkonzentration der verdünnten Stammlösung berechnet. 5 ml der 250 ppm Stammlösungen wurden auf 50 ml verdünnt. Bei ppm handelt es sich um eine Massenkonzentration, da 1 ppm= 1μg/ml. Aus diesem Grund können wir die Formel für Verdünnungen von Massenkonzentrationen anwenden:
⚠ $$ \omega_{1} = \frac {\omega_{2} \cdot V_{2}} {V_{1}}⚠ $$
- ω1 = Massenkonzentration der verdünnten Stammlösung [ ppm] Ergebnis: 25ppm
- ω2 = Massenkonzentration der unverdünnten Stammlösung [ ppm] hier : 250ppm
- V2 = Volumen [ ml] hier : 5m
- V2 = Volumen [ ml] hier : 50ml
- Berechnung Halbstufenpotentiale
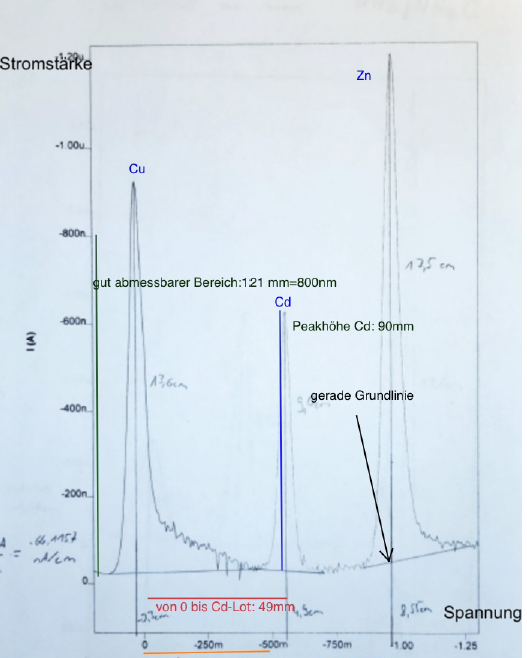
Bei der Berechnung der Halbstufenpotentiale muss auf der x-Achse die Länge eines beliebigen Voltbereiches ausgemessen werden. In der Abbildung 5 kann man beispielhaft sehen, dass der Bereich von 0 bis 500 mV 44 mm lang ist. Durch Dreisatz wird dann ausgerechnet wie viel Volt ein Millimeter sind (Maßstab: 1mm entspricht 11,363636mV). Anschließend wird der Abstand von 0 Volt bis zum Lot der Analyten ausgemessen (Abbildung 6: der Abstand von 0 V bis zum Cd-Lot ist 49 mm lang). Das Halbstufenpotential kann dann nach folgender Gleichung berechnet werden:
⚠ $$E_{1/2} = Abstand_{x-Achse} \cdot Maßstab⚠ $$
- E1/2 = Halbstufenpotential [V]
- Abstandx-Achse = Abstand von 0 V bis Lot [mm]
- Maßstab = wie viel mV einem mm der x-Achse entsprechen [mV / 1mm]
- Berechnung Massenanteile der Analyten (Cu,Cd,Zn)
Zunächst wird bei dem Polarogramm der Standardlösung und der Probe der Maßstab bestimmt. Der Maßstab wird erhalten, indem an der y-Achse ausgemessen wird, wie lang ein bestimmter Amperebereich ist (Abbildung 6: der Bereich von 0 ist 800 nA ist 121 mm lang). Über den Dreisatz berechnet man dann, wie viel Ampere einem Millimeter entsprechen (Beispiel 1mm entspricht 6,611570248 nA). Es muss dabei beachtet werden, dass der y-Achsen Abschnitt der Standardlösung und der Probe nicht den gleichen Präfix bei der Einheit haben. Die Maßstäbe können in Mikroampere oder Nanoampere angegeben werden. Danach wird bei dem Polarogramm der Standardlösung und der Probe die Höhe des Peaks von der Grundlinie aus abgemessen (in der Abbildung 6 ist der Cd-Peak 90mm lang).
Zuletzt werden die Werte in die Formel eingesetzt.
⚠ $$ \omega = \frac {25ppm \cdot h_{Peak Probe} \cdot Maßstab_{Probe} }{h_{Peak Standard} \cdot Maßstab_{Standard} } ⚠ $$
- 25 ppm wurde im 1.Schritt berechnet
- hPeak Probe = Peakhöhe der Probenlösung [mm]
- MaßstabProbe = wie viel nA 1mm der y-Achse entsprechen [na/1mm]
- hPeak Standard = Peakhöhe der Standardlösung [mm]
- MaßstabStandard = wie viel nA 1mm der y-Achse entsprechen [nA/1mm]
Einzelnachweise
1 Eberhard Ehlers: „Analytik II-Kurzlehrbuch", 12.Auflage S.308 ⇑
2 https://www.ipc.uni-jena.de/ipcmedia/lehre/iaii/iaii_02_polarographie.pdf (Letzter Stand: 1.12.2021) ⇑
3 Rücker/Neugebauer/Willems: „Instrumentelle pharmazeutische Analytik", Wissenschaftliche Vertragsgesellschaft Stuttgart, 5.Auflage, S.635 ⇑
4 Dominik Steinhilber Wurglics: „Instrumentelle Analytik kompakt", Wissenschaftliche Verlagsgesellschaft Stuttgart, 3.Auflage ⇑
5 Sergio Petrozzi: „Instrumentelle Analytik", WILEY-VCH,1.Auflage, S.193-203 ⇑
6 Sergio Petrozzi: „Instrumentelle Analytik", WILEY-VCH, 1.Auflage, S.193-203 ⇑
7 Daniel C.Harris: „Lehrbuch der Quantitativen Analyse", 8.Auflage, S.428 ⇑
8 Sergio Petrozzi: „Instrumentelle Analytik", WILEY-VCH, 1.Auflage, S.193-203 ⇑
9 "Elektrochemische Spannungsreihe" https://www.chemie.de/lexikon/Elektrochemische_Spannungsreihe.html (Letzter Stand: 2.12.2021) ⇑
10 Dominik Steinhilber Wurglics:„Instrumentelle Analytik kompakt“, Wissenschaftliche Verlagsgesellschaft Stuttgart, 3.Auflage ⇑
11 Sergio Petrozzi: „Instumentelle Analytik“, WILEY-VCH, 1.Auflage, S.193-203 ⇑
12 Dominik Steinhilber Wurglics: „Instrumentelle Analytik kompakt“, Wissenschaftliche Verlagsgesellschaft Stuttgart, 3.Auflage ⇑
13 von Mahdia Djawadi gezeichnet ⇑
14 von Mahdia Djawadi gezeichnet ⇑
15 "Elektrochemische Spannungsreihe“ https://www.chemie.de/lexikon/Elektrochemische_Spannungsreihe.html (Letzter Stand: 2.12.2021) ⇑
16 von Annabell Steinke gezeichnet ⇑
17 Foto:Iman Bischo ⇑
18 Sergio Petrozzi: „Instumentelle Analytik“, WILEY-VCH, 1.Auflage, S.193-203 ⇑
19 „Polarographie“ https://www.ipc.uni-jena.de/ipcmedia/lehre/iaii/iaii_02_polarographie.pdf (Letzter Stand: 1.12.2021) ⇑
20 Praktikumsskript:„Instrumentelle Analytik", 4.Semester, Dr. Thomas Kellner, WiSe 2021/22, S.29 ⇑
21 Dominik Steinhilber Wurglics: „Instrumentelle Analytik kompakt“, Wissenschaftliche Verlagsgesellschaft Stuttgart, 3.Auflage ⇑
22 Sergio Petrozzi: „Instumentelle Analytik“, WILEY-VCH, 1.Auflage, S.193-203 ⇑
23 Rücker/Neugebauer/Willems: „Instrumentelle pharmazeutische Analytik", Wissenschaftliche Vertragsgesellschaft Stuttgart, 5.Auflage, S.635 ⇑
24 Dominik Steinhilber Wurglics: „Instrumentelle Analytik kompakt“, Wissenschaftliche Verlagsgesellschaft Stuttgart, 3.Auflage ⇑
25 Skizze aus Praktikumsprotokoll von Annabell Steinke ⇑
