Konduktometrie
Titelblatt
Bericht der Expert*innengruppe für
Konduktometrie
SoSe 2021
Abgabedatum
21.06.2021
Über-/Expert*innengruppe 02
Leonie Funke
Vivien Grieb
Andre Lakeit
Anna Leis
Janine Schmidt
Malte Theile
Konduktometrie
Inhaltsverzeichnis
Einleitung
Die Konduktometrie oder auch konduktometrische Titration ist eine auf der elektrischen Leitfähigkeit unterschiedlicher Ionen basierende elektrochemische Analysetechnik. Sie kann zur Bestimmung des Elektrolytgehaltes von Elektrolytlösungen (Absolutmessung) und zur Erkennung des Endpunktes bei Titrationen verwendet werden (konduktometrische Indizierung).
Für die elektrische Leitfähigkeit sind nur die dissoziierten Ionen und deren Konzentrationen bzw. Konzentrationsänderungen relevant, weshalb in der Konduktometrie nur Reaktionen qualitativ und quantitativ bestimmt werden können, bei denen sich entweder die Zahl der Ladungsträger verändert oder Ionen durch andere Ionen mit stark abweichender Ionenleitfähigkeit ersetzt werden.
Die Konduktometrie kann bei Säure-Base-Titrationen, komplexometrischen Titrationen und Fällungstitrationen, sowie bei der Indizierung im nicht-wässrigen Medium und bei Simultanbestimmungen verwendet werden. Hierbei zeichnet sich die konduktometrische Titration vor allem dadurch aus, dass die Konzentration des Titranden geringer sein kann als bei anderen Analyseverfahren. Dadurch ist die Konduktometrie besonders gut bei kleinen Analysenmengen einsetzbar.
Analyseverfahren, bei denen die Konduktometrie nicht oder nur selten verwendet werden kann, sind Redoxtitrationen, da die Leitfähigkeiten der Ionenarten sich meist nicht genug voneinander unterscheiden lassen, um eine geeignete Aussage über einen Titrationsverlauf machen zu können. Zusätzlich können im sauren Milieu Matrixeffekte auftreten, welche den Äquivalenzpunkt verschleiern können.1
Im Europäischen Arzneibuch wird die Konduktometrie zur Reinheitsprüfung von Medien gebraucht, unter anderem zur mikrobiologischen Überwachung bei Wasser für Injektionszwecke. 2
Chemische und physikalische Grundlagen
Wässrige Lösungen von Elektrolyten leiten durch die Bewegung von Ladungsträgern den elektrischen Strom. Die Ionenleitung kann dabei durch Diffusion, Konvektion oder Migration (Ionen-Wanderung im elektrischen Feld) erfolgen. Bei der Konduktometrie wird die elektrische Leitfähigkeit von Elektrolytlösungen, die auf der Migration beruht, herangezogen. Zur Leitfähigkeitsmessung wird der elektrische Widerstand R bzw. der Leitwert G (in ⚠ $ S = \tfrac{1}{Ω} $) bestimmt.3 Es gilt:
⚠ $$ G= \tfrac{1}{R} ⚠ $$
Mithilfe des Ohm‘schen Gesetzes
⚠ $$ R= \tfrac{U}{I} ⚠ $$
lässt sich aus der Spannung U und dem Strom I der elektrische Widerstand R für einen metallischen elektrischen Leiter bestimmen.
Bei der Konduktometrie wird allerdings mit Elektrolytlösungen gearbeitet, für die nicht das Ohm’sche Gesetz gilt, da die Polarisationsspannung ⚠ $ U_P $ mitberücksichtigt werden muss. Polarisation beschreibt den Zustand, bei dem es trotz anliegender Spannung keinen Stromfluss gibt. Der Stromfluss stagniert, da sich durch die Elektrolyse (ohne Umrühren) um die Anode eine Überschuss- und um die Kathode eine Verarmungszone gebildet hat. Es gilt für den elektrischen Widerstand R:
⚠ $$ R= \tfrac{U \ - \ U_P}{I} ⚠ $$
Das Auftreten der Polarisationsspannung muss verhindert werden, da diese nicht messbar ist und somit eine fehlerfreie Messung des Widerstandes und damit des Leitwerts nicht möglich wäre. Daher erfolgt die Messung, im Gegensatz zu allen anderen elektrochemischen Methoden, mit Wechselspannung mit hoher Frequenz. Diese verhindert durch den ständigen Polaritätswechsel den Aufbau einer Polarisationsspannung. Außerdem verwendet man platinierte Elektroden, die aufgrund ihrer größeren Oberfläche schwerer polarisiert werden können.
Der Widerstand eines Leiters R (in ⚠ $ Ω = \tfrac{1}{S} $) kann gemäß
⚠ $$ R= ρ\cdot\tfrac{L}{S} ⚠ $$
aus dem spezifischen Widerstand ρ, der Länge L und dem Querschnitt S ermittelt werden. Der Widerstand einer Lösung wird geringer, wenn die Elektrodenoberfläche des Messgerätes (S) zunimmt oder der Abstand zwischen den Elektroden (L) abnimmt.
Der Kehrwert des spezifischen Widerstandes ρ wird als Leitfähigkeit κ (in ⚠ $ \tfrac{S}{cm} $) bezeichnet:
⚠ $$ κ = \tfrac{1}{ρ} ⚠ $$
Die Leitfähigkeit κ lässt sich nach
⚠ $$ κ = \tfrac{1}{ρ} = \tfrac{1}{R} \cdot \tfrac{L}{S} = G \cdot K_{Zelle} ⚠ $$
direkt aus dem Leitwert G und der Zellkonstante ⚠ $ K_{Zelle}$ der Leitfähigkeitszelle berechnen. Durch ⚠ $ K_{Zelle} = \tfrac{L}{S}$ werden Abweichungen der verwendeten Leitfähigkeitszelle von einer idealen Leitfähigkeitszelle mit dem Volumen von ⚠ ${1 cm \ \cdot 1 cm \ \cdot 1 cm = 1 cm^3}$ berücksichtigt.
Zu beachten ist, dass die Leitfähigkeit und damit der Leitwert nur bei verdünnten Lösungen (mit Konzentration c < 0,1 mol/L) und konstanter Temperatur proportional zur Ionenkonzentration ist. Erhöhte Temperaturen führen aufgrund der größeren Wechselwirkungen zu einem hohen Dissoziationsgrad, sodass die Proportionalität von Temperatur und Ionenkonzentration nicht mehr gegeben ist. Im Gegensatz dazu liegen Teilchen bei hohen Konzentrationen nicht vollständig dissoziiert vor. Dies ist problematisch, da für die Ionenleitung nur dissoziierte Teilchen relevant sind. Die Leitfähigkeit ist also umso größer, je mehr frei bewegliche Ionen in der Lösung vorhanden sind. Dabei sollte beachtet werden, dass die Solvathülle die Ionenbeweglichkeit deutlich herabsetzt.
Für eine bessere Vergleichbarkeit lässt sich aus der Leitfähigkeit κ die molare Leitfähigkeit Λ (in ⚠ $ \tfrac{S \ \cdot \ cm^2}{mol} $) mithilfe folgender Formel berechnen:
⚠ $$ Λ = \tfrac{1000 \ \cdot \ κ}{c} ⚠ $$
Der Faktor 1000 ergibt sich aus der Umwandlung der Einheit von c (in ⚠ $ \tfrac{mol}{L} $): ⚠ $ 1 L = 1000 \ cm^3 $.
Die Äquivalentleitfähigkeit Λ* (in ⚠ $ \tfrac{S \ \cdot \ cm^2}{mol} $) bezieht zusätzlich die Ladungszahl z und den stöchiometrischen Koeffizienten n im Elektrolytmolekül in die Berechnung ein:
⚠ $$ Λ^* = \tfrac{1000 \ \cdot \ κ}{c \ \cdot\ z \ \cdot\ n} ⚠ $$
Das Leitvermögen ist abhängig von der Konzentration c und der Ladungszahl z der Ionen. Je höher die Verdünnung, desto größer ist die molare Leitfähigkeit bzw. Äquivalentleitfähigkeit, denn die molare Leitfähigkeit nimmt bei zu hoher Ionenkonzentration aufgrund von Abstoßungskräften ab. Für starke Elektrolyte (z.B. HCl) gilt das Kohlrau’sche Quadratwurzelgesetz:
⚠ $$ Λ = Λ∞ - k \cdot \sqrt{c} ⚠ $$
Der Proportionalitätsfaktor k wird allgemein mit der Ionenladung größer. Bei unendlicher Verdünnung gilt:
⚠ $$ Λ = Λ∞ ⚠ $$
Die molare Grenzleitfähigkeit Λ∞ (in ⚠ $ \tfrac{S \ \cdot \ cm^2}{mol} $) entspricht somit der molaren Leitfähigkeit Λ.
Die molare Grenzleitfähigkeit eines Salzes lässt sich aus den Grenzionenäquivalentleitfähigkeiten Λ+,∞ (in ⚠ $ \tfrac{S \ \cdot \ cm^2}{mol} $) und Λ-,∞ (in ⚠ $ \tfrac{S \ \cdot \ cm^2}{mol} $) seiner Anionen und Kationen sowie den stöchiometrischen Faktoren n+ und n- wie folgt berechnen:
⚠ $$ Λ∞ = n_+ \cdot Λ_{+,∞} + n_- \cdot Λ_{-,∞} ⚠ $$
Die gesamte Grenzleitfähigkeit ist also die Summe der Teilleitfähigkeiten.
Die Grenzionenäquivalentleitfähigkeit (= Grenzleitfähigkeit) ist ein Maß für die Beweglichkeit bzw. Wanderungsgeschwindigkeit der Ionen bei unendlicher Verdünnung, die sicherstellt, dass es nicht zu Wechselwirkungen zwischen den Ionen kommt. Da dies eine ideale Lösung ist, sind die tabellierten Grenzionenäquivalentleitfähigkeiten entsprechend auch nur ideale Werte.
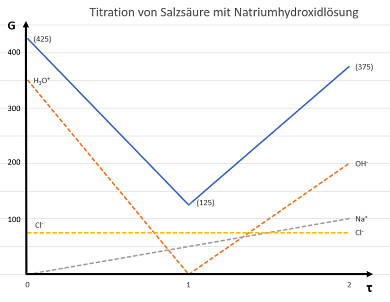
Das Leitvermögen ist also nicht nur abhängig von der Konzentration c und der Ladungszahl z der Ionen, sondern auch von der Wanderungsgeschwindigkeit v oder der Ionenbeweglichkeit u.
Bei 25 °C in Wasser haben Oxonium-Ionen mit etwa ⚠ $ 350 \tfrac{S \ \cdot \ cm^2}{mol} $ und Hydroxid-Ionen mit knapp ⚠ $ 200 \tfrac{S \ \cdot \ cm^2}{mol} $ außergewöhnlich hohe Grenzleitfähigkeiten aufgrund des sogenannten Tunneleffekts. Dieser Effekt beschreibt, dass Oxonium- und Hydroxid-Ionen die Ladung über Wasserstoffbrückenketten von Teilchen zu Teilchen weiterleiten, weshalb sie in der Lage sind, den Strom besonders schnell und effektiv zu leiten. Im Gegensatz dazu wandern alle anderen Ionen selber durch die Lösung, was durch die die Ionen umgebende Solvathülle erschwert wird.
Mithilfe der Grenzleitfähigkeiten lässt sich der Verlauf der Titrationskurve vorhersagen, wie auf der Abbildung zu erkennen ist. Hierbei handelt es sich um die Titration von Salzsäure mit Natriumhydroxid-Lösung bei konduktometrischer Indikation. Aufgrund der besonders hohen Grenzleitfähigkeiten der Oxonium- und Hydroxidionen lässt sich der Titrationsendpunkt dieser Säure-Base-Titration sehr gut mittels Konduktometrie bestimmen. Vor dem Äquivalenzpunkt ist die Leitfähigkeit aufgrund der Oxoniumionen hoch; sie sinkt aber mit Fortschreiten der Titration bis zum Äquivalenzpunkt bei τ = 1 durch H2O-Entstehung aus den H3O+- und OH--Ionen. Trotzdem ist am Äquivalenzpunkt durch Na+- und Cl--Ionen, deren Grenzionenäquivalentleitfähigkeiten sich addieren, noch eine Leitfähigkeit messbar. Die steigende Leitfähigkeit nach dem Äquivalenzpunkt ist auf den Überschuss an Natriumhydroxid-Maßlösung zurückzuführen.5 6
Instrumenteller Aufbau
Einfache konduktometrische Messung
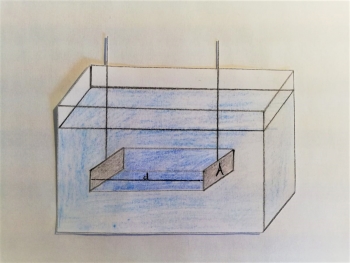
Einfacher Aufbau eines Konduktometers mit zwei zueinander parallelen Elektroden in der zu prüfenden Lösung.
Der Aufbau einer konduktometrischen Messung besteht aus der zu untersuchenden Lösung, welche in einem Glasgefäß vorliegt, sowie einer Messeinheit, welche über zwei sich in der Lösung befindlichen inerten Elektroden Messwerte zur elektrischen Leitfähigkeit der Lösung ermittelt. Die Elektroden sind parallel zueinander ausgerichtet und sollten so weit in die Probenlösung eingetaucht werden, dass die oberen Öffnungen mit Flüssigkeit bedeckt sind. Eine alternative Vorrichtung stellt ein Glasgefäß dar, in dem die Elektroden nicht von oben in die Lösung eingetaucht werden, sondern direkt in das Gefäß selbst eingeschmolzen sind und sich in diesem gegenüber stehen.7 Sie bestehen meist aus Platinblechen, deren Oberfläche zur Vergrößerung mit Platinschwarz überzogen ist, um einer Polarisation entgegenzuwirken.8 Da die Bleche oft per Hand angefertigt werden entstehen leichte Abweichungen, welche in der Messung durch die Zellkonstante ⚠ $ K_{Zelle}$ ausgeglichen werden.9 Zudem umschließen die Elektroden idealerweise einen Wasserwürfel mit einer Kantenlänge von 1cm (in der Abb. als "d" gekennzeichnet).10
Zur Messung wird eine Wechselspannung angelegt, da eine Überspannung bzw. Polarisation der Elektroden und damit eine Messabweichung vermieden werden soll.11 Dazu wechseln die Elektroden die Polung hin und her, sodass sich keine Ansammlung von Ionen bilden kann. Bei Gleichstrom würde diese Schicht aus polarisierten Ionen an den Elektroden entstehen, wodurch der Widerstand steigen und der Leitwert mit der Zeit gen Null sinken würde.12
Konduktometrische Titration
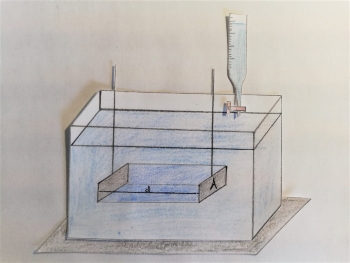
Aufbau einer Titration mit Konduktometer, Magnetrührer unterhalb und einer mit Maßlösung gefüllten Bürette.
Bei der konduktometrischen Titration wird derselbe Aufbau wie bei der normalen konduktometrischen Messung verwendet. Ergänzt wird der Aufbau durch eine Bürette, über die die bei der Titration verwendete Maßlösung zugegeben werden kann. Zusätzlich begünstigt ein unter dem Glasgefäß stehender Magnetrührer die schnelle Verteilung der Maßlösung (Konvektionsströmung) und damit die Reaktionsgeschwindigkeit.13
Allgemein müssen die zu titrierenden Substanzen eine gute Leitfähigkeit besitzen, wodurch die Anwendung auf Säure-Base-, Fällungs- und Komplexbildungstitrationen beschränkt ist. Die Maßlösung sollte möglichst hoch konzentriert sein, um eine Volumenvergrößerung zu vermeiden, welche wiederum die Grenzionenäquivalentleitfähigkeit beeinflussen würde.14
Einzelnachweise
1 Rücker et al., Instrumentelle pharmazeutische Analytik, 5. Auflage, 2013, Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 664 ⇑
4 nach: Dr. Oliver Orban, Skript zur "Einführung in die Instrumentelle Analytik: Elektrochemische Methoden", TU Braunschweig, WiSe 2020, S. 19 ⇑
5 Rücker et al., Instrumentelle pharmazeutische Analytik, 5. Auflage, 2013, Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 546-550 und S. 664-668 ⇑
6 Dr. Oliver Orban, Skript zur "Einführung in die Instrumentelle Analytik: Elektrochemische Methoden", TU Braunschweig, WiSe 2020, S. 1-22 ⇑
7 Rücker et al., Instrumentelle pharmazeutische Analytik, 5. Auflage, 2013, Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 666/667 ⇑
8 Rücker et al., Instrumentelle pharmazeutische Analytik, 5. Auflage, 2013, Wissenschaftliche Verlagsgesellschaft Stuttgart, S. 666/667 ⇑
10 Dr. Oliver Orban, Skript zur "Einführung in die Instrumentelle Analytik: Elektrochemische Methoden", TU Braunschweig, WiSe 2020, S. 1-22 ⇑
12 Dr. Oliver Orban, Skript zur "Einführung in die Instrumentelle Analytik: Elektrochemische Methoden", TU Braunschweig, WiSe 2020, S. 1-22 ⇑
13 Dr. Oliver Orban, Skript zur "Einführung in die Instrumentelle Analytik: Elektrochemische Methoden", TU Braunschweig, WiSe 2020, S. 1-22 ⇑
Monographie: Voriconazol
Stoffcharakterisierung (Wirkung und Anwendung)
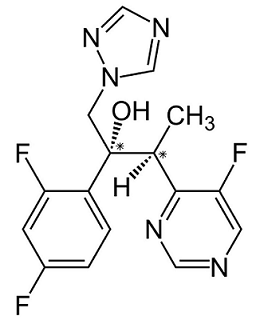
Voriconazol ist ein Breitspektrum-Antimykotikum aus der Gruppe der Triazol-Antimykotika, die als gemeinsames Merkmal eine Triazol-Ringstruktur aufweisen. Die Bezeichnung nach IUPAC lautet (2R,3S)-2-(2,4-Difluorphenyl)-3-(5-fluorpyrimidin-4-yl)-1-(1H-1,2,4-triazol-1-yl)butan-2-ol und die Summenformel C16H14F3N5O. Das Molekulargewicht beträgt 349,3 g/mol. 2 Es besitzt 2 Chiralitätszentren, die in der Abbildung markiert sind. Von den 4 möglichen Diastereomeren weist fast ausschließlich das (2R,3S)-Isomer (Voriconazol) eine starke antimykotische Wirkung auf. Das europäische Arzneibuch lässt daher auf Verunreinigungen durch die drei unerwünschten Diastereomere prüfen.
Voriconazol kann als weißes bis fast weißes Pulver klassifiziert werden, welches sehr schwer löslich in Wasser, aber leicht löslich in Aceton und Dichlormethan ist. Es ist in Form von Filmtabletten, als Pulver zur Herstellung einer Infusionslösung und als Pulver zur Herstellung einer Suspension im Handel. In fester Form gilt die Substanz als relativ stabil. Der Wirkstoff ist empfindlich gegenüber UV-Licht, Feuchtigkeit und bei pH-Wert < 6 spaltet sich der Wirkstoff zu achiralen Produkten auf, die den Edukten der Synthese entsprechen. 3
Eingesetzt wird Voriconazol bei lokalen und systemischen Pilzinfektionen, die durch die Krankheitserreger der Gattungen Aspergillus, Scedosporium und Fusarium hervorgerufen werden. Es dient außerdem zur Therapie von invasiven Candida-Infektionen. Da es liquorgängig ist, kann es auch gegen zentrale Mykosen eingesetzt werden.4 Die fungistatische Wirkung beruht auf der Hemmung der Ergosterolbiosynthese der Pilze. Ergosterol ist ein essentieller Bestandteil der Zellmembran von Pilzen und sorgt für Stabilität und Flexibilität. Somit ist die Membran widerstandsfähig und der Zellinhalt bleibt dadurch geschützt.5 Durch den Wirkstoff kommt es nun dazu, dass die CYP450-abhängige Lanosterol-14α-Demethylase gehemmt wird. CYP450 sind Hämproteine mit enzymatischer Aktivität und dienen der Oxidation vieler körpereigener und körperfremder Substanzen. Durch die Hemmung der Lanosterol-14α-Demethylase erfolgt keine Umsetzung mehr von Lanosterin (Zwischenprodukt bei der Biosynthese) zu Ergosterin. Deshalb bilden sich Einlagerungen falscher Sterole in der Membran, wodurch die normale Funktion der Membran gestört wird. Dies wirkt sich vor allem auf die Funktion der membranständigen Enzyme aus. Als Beispiel wäre die Chitinsynthethase zu nennen, die für das Zellwachstum und die Teilung von Pilzen unverzichtbar ist. Zusätzlich wirkt Voriconazol auch fungizid, da es sich selbst in die Zellmembran einlagern kann und derartige Strukturveränderungen mit sich bringt, dass Zellbestandteile austreten können. 6
Durchführung der Monographie
Die Monographie zu Voriconazol im Europäischen Arzneibuch sieht unter anderem eine Prüfung auf mögliche Kontamination mit Camphersulfonsäure, der sogenannten „Verunreinigung E“, vor. Da nur hier die Konduktometrie Verwendung findet, beschränken sich die folgenden Abschnitte auch nur darauf. Der eigentliche Nachweis von Verunreinigung E erfolgt dabei durch Flüssigchromatographie, nur die anschließende Detektion verläuft konduktometrisch.
Zunächst sind die mobile Phase, sowie die Untersuchungslösung und drei Referenzlösungen frisch herzustellen. Zur Herstellung der mobilen Phase wird demineralisiertes Wasser R mit Methanol R vermischt und es wird eine Natriumhydroxidlösung zugesetzt. Für die Untersuchungslösung wird die zu prüfende Substanz in Methanol R gelöst – anschließend wird mit der mobilen Phase aufgefüllt. Referenzlösung a besteht aus der nachzuweisenden Verunreinigung E und auch hier wird die mobile Phase zugesetzt. Durch weitere Verdünnung dieser Lösung erhält man die Referenzlösung c – nur diese wird bei der späteren Durchführung auch verwendet und Referenzlösung a wird verworfen. Referenzlösung b dient zur Prüfung der Trennleistung und enthält Chlorid-Ionen. Als stationäre Phase ist ein stark basischer Anionenaustauscher zur Chromatographie R vorgeschrieben.
Nacheinander werden von der Untersuchungslösung, dann von der Referenzlösung b und anschließend von der Referenzlösung c jeweils 20 μL in die Säule eingespritzt. Zur Detektion tropfen die getrennten Komponenten in eine konduktometrische Apparatur – die Detektion verläuft also über die Leitfähigkeit der Bestandteile, wodurch auf die Konzentration Rückschlüsse gezogen werden können. Zur Verbesserung der Detektierbarkeit passieren die Untersuchungslösungen zuvor einen Anionensuppressor. 7 8
Auswertung/ Interpretation/ Bedeutung und Eignung der analytischen Methode
Eigenschaften der Verunreinigung E
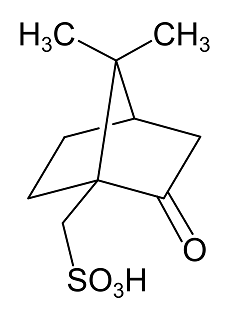
Bei der Verunreinigung E handelt es sich um [(1R,4S)-7,7-Dimethyl-2-oxobicyclo[2.2.1]hept-1-yl]methansulfonsäure.9 Sie wird im Herstellungsprozess des Voriconazols zur Racemattrennung eingesetzt und kann daher in Rückständen in der Substanz enthalten sein.10 Voraussetzung für die Anwendung einer Ionenaustauschchromatographie zur Trennung und eines Leitfähigkeitsdetektors zur anschließenden Detektion der Verunreinigung E ist ihr Vorliegen als Anion. Betrachtet man die abgebildete Struktur, so lässt sich dies leicht durch die enthaltene stark saure Sulfonsäuregruppe mit pKs < 1 erklären.11 Es ist davon auszugehen, dass die Verunreinigung schon in neutraler Lösung vollständig dissoziiert vorliegt.
Chromatographische Trennung
In der chromatographischen Säule wird die Verunreinigung E aufgrund von ionischen Wechselwirkungen mit der stationären Phase stärker zurückgehalten als andere, nicht geladene Komponenten wie beispielsweise das Voriconazol. Dadurch erreicht sie nach einer längeren, charakteristischen relativen Retentionszeit den Detektor.
Detektion
Der Leitfähigkeitsdetektor funktioniert nach dem oben beschriebenen Prinzip der Konduktometrie. Gelangen zusätzliche ladungstransportierende Teilchen in Form der Verunreinigung E in den Detektor, so kommt es zum erhöhten Stromfluss zwischen den beiden Elektroden. Aus diesem Anstieg lässt sich die Leitfähigkeit ermitteln, welche sich bei Analytkonzentrationen von unter 0,1 mol/l proportional zur Konzentration verhält. Für die Berechnungen wird die Zellkonstante benötigt. Diese kann durch Messung mit NaCl-Lösung (Referenz b) ermittelt werden.13
Auswertung des Chromatogramms
Im vom Detektor gelieferten Chromatogramm erscheint ein Peak, dessen Fläche proportional zur Konzentration der Verunreinigung E ist. Um eine Aussage darüber zu erhalten, ob der durch das Arzneibuch vorgeschriebene Grenzwert eingehalten wurde, wird ein Vergleich mit dem Chromatogramm der Referenzlösung c durchgeführt. Diese enthält eine definierte Menge der Verunreinigung E als Hauptkomponente. Die Fläche des zur Verunreinigung E gehörenden Peaks der Untersuchungslösung darf die Fläche des Hauptpeaks im Chromatogramm der Referenzlösung c nicht überschreiten. Dies entspricht einem Anteil von 0,10 Prozent an der Gesamtsubstanz. Um sicherzustellen, dass das erhaltene Chromatogramm zur Auswertung geeignet ist und verlässliche Aussagen liefert, werden zusätzlich die Auflösung und der Symmetriefaktor überprüft. Ein hinreichender Wert für die Auflösung bedeutet, dass die Peaks nacheinander eluierender Komponenten im Chromatogramm ausreichend gut voneinander getrennt erscheinen. Dies wird hier anhand der Referenzlösung b überprüft. Die Auflösung zwischen den Peaks von Verunreinigung E und Chlorid sollte mindestens 3,5 betragen. Ein ausreichend kleiner Symmetriefaktor ist notwendig, damit die für die Berechnungen verwendeten Modellannahmen, zum Beispiel zur Peakform, als gültig betrachtet werden können.14 Zur Ermittlung wird der Peak der Verunreinigung E im Chromatogramm der Referenzlösung c verwendet. Der Symmetriefaktor sollte hierbei einen Wert von 1,7 nicht überschreiten.
Bedeutung und Eignung der Methode
Die dargestellte Methode eignet sich prinzipiell für die Trennung jeglicher ionischer Komponenten, die nach jeweils unterschiedlich langer Retention in der chromatographischen Säule durch den Leitfähigkeitsdetektor erkannt werden können. Sie hat damit eine hohe Bedeutung für viele Anwendungsgebiete, darunter die Untersuchung und Reinigung von Trinkwasser sowie die Trennung von Proteinen.15 Insbesondere für Reinheitsprüfungen wird sie häufig empfohlen.
Einzelnachweise
5 Informationsdienst Wissenschaft/Wirkungsweise von Pilz-Medikamenten, zuletzt abgerufen am 10.05.21 um 20:03 ⇑
6 Pharmazeutische Zeitung/Azol-Antimykotikum Voriconazol, zuletzt abgerufen am 15.06.2021 um 14:49 ⇑
TU-Braunschweig Institut für Medizinische und Pharmazeutische Chemie Seminar: Instrumentelle Analytik Kontakt: tubs@t-kellner.de
