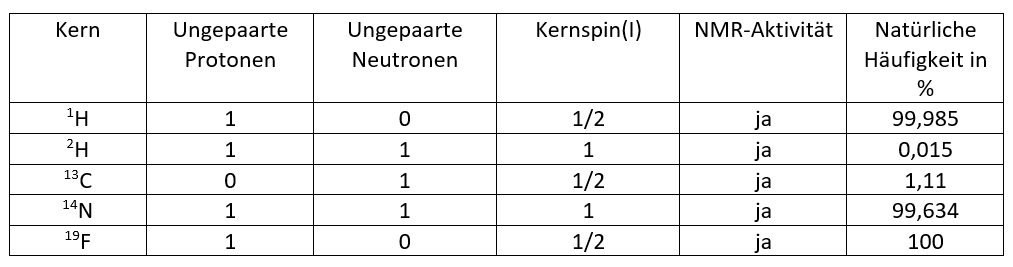
Kernresonanzspektroskopie
Titelblatt
Bericht der Expertengruppe für
Kernresonanzspektroskopie
SoSe 2021
Abgabedatum
21.06.2021
Über-/Expertengruppe 08
Fatma Gül Özyilmaz
Sahar Ander
Christian Krahtz
Madlen Herbst
Delia Bröker
Gina Heyen
NMR-Spektroskopie
Inhaltsverzeichnis
Einleitung
Die NMR-Spektroskopie (engl.: nuclear magnetic resonance spectroscopy), auch Kernresonanzspektroskopie genannt, ist eine der wichtigsten analytischen Methoden zur Strukturaufklärung organischer Verbindungen. Sie beruht auf dem unterschiedlichen Verhalten magnetisch aktiver Atomkerne in einem extern angelegten, starken Magnetfeld.
Im Folgenden werden die 1H-NMR-Spektroskopie und die 13C-NMR-Spektroskopie aufgegriffen. Bei beiden Methoden werden Radiowellen mit einer Wellenlänge von über einem Dezimeter zur Bestrahlung von Atomkernen verwendet. Diese bewirken, dass die Orientierungen der Kernspins der Atome in einem äußeren Magnetfeld umgekehrt werden, wodurch diese Atome auf unterschiedliche Energieniveaus gelangen, welche vermessen und analysiert werden.
Das NMR-Spektrum gibt Aufschluss über die Anzahl der Atome, über ihre Verknüpfung in einem Molekül sowie Informationen über die Umgebung des zu analysierenden Atomkerns. 1 2 3
Physikalische und chemische Grundlagen der NMR-Spektroskopie
Kernspin und magnetisches Moment von Atomkernen
Kerne besitzen, neben ihrer Ladung und Masse, einen Drehimpuls um ihre eigene Achse. Dieser Eigendrehimpuls bzw. diese Rotation der Kernladung wird als Kernspin bezeichnet. Da die Kerne eine positive elektrische Ladung haben und rotieren, erzeugen diese ein Magnetfeld. Aus dem Grund besitzen alle Atomkerne mit einem Drehimpuls auch ein magnetisches Moment. Das durch die Rotation der Kernladung entstandene Magnetfeld, kann man sich vereinfacht als einen Stabmagneten vorstellen. Die Richtung des Magnetfelds folgt hierbei der Drehrichtung des Kerns.
Vergleicht man nun die Drehrichtung der Atomkerne, können diese zwei unterschiedliche Orientierungen einnehmen, welche dann entweder parallel (↑↑) oder antiparallel (↑↓) zum Magnetfeld ausgerichtet sind. Diese zwei Spinrichtungen besitzen energetisch unterschiedliche Zustände.
Entsprechend der Quantentheorie ist nur eine begrenzte Anzahl von Orientierungsmöglichkeiten möglich, da sich ein Atom nicht beliebig in einem Magnetfeld drehen kann. Die Anzahl dieser Orientierungsmöglichkeiten werden durch die Kernspinquantenzahl (I) definiert.
Hierbei kann diese halbzahlige oder ganzzahlige Werte annehmen. Der Gesamtspin eines Atoms ist abhängig vom Kernaufbau und ergibt sich aus der Summe der enthaltenen Kernbausteine, d.h. der Protonen und Neutronen. Daraus ergeben sich folgende Gesetzmäßigkeiten:
Atomkerne mit einer geraden Massen- und Ordnungszahl (g/g-Kerne) sind magnetisch nicht aktiv und somit für die Kernresonanzmessung nicht zugänglich, da sich die Spins gegenseitig aufheben. Die Kernspinquantenzahl entspricht dann I = 0. Dies ist beispielsweise beim 12 C-Atom der Fall.
Sind Massenzahl oder Ordnungszahl oder beide Zahlen ungerade (u/u-, u/g-, g/u-Kerne), dann sind diese Kerne der NMR-Spektroskopie zugänglich. 4 5 6 7
Präzessionsbewegung
Ergänzend zur Drehung um die Längsachse, existiert auch, die durch das Anstoßen des Kerns von der Seite bedingte, Präzessionsbewegung, welche in der 1H-NMR-Spektroskopie durch ein extern angelegtes, starkes, homogenes und statisches Magnetfeld induziert wird. Diese Bewegung gleicht der torkelnden Bewegung eines mechanischen Kreisels. Die Kreisfrequenz ⚠ $w_0$ der Torkelbewegung (synonym zu Präzessionsbewegung) wird durch die Larmor-Gleichung beschrieben. ⚠ $$ w_0 = 2⋅π⋅v = y⋅B_0 ⚠ $$
⚠ $w_0$: Kreisfrequenz [1/s = Hz]
⚠ $v$: Frequenz [1/s = Hz]
⚠ $y$: magnetogyrisches Verhältnis (MHz/Tesla); Proportionalitätskonstante: spezifisch für jedes Isotop
⚠ $B_0$: Stärke des äußeren Magnetfelds (Tesla)
Es wird deutlich, dass die Kreisfrequenz der Präzessionsbewegung zunimmt, je stärker das angelegte Magnetfeld ist. Hierbei kann die Präzessionsbewegung gleichgerichtet (α-Spin) zum angelegten Magnetfeld oder entgegengesetzt (β-Spin) sein. Bei dem gleichgerichteten Magnetfeld handelt es sich um den energetisch günstigeren bzw. um den energieärmeren Zustand. Daher ist dieser Zustand wahrscheinlicher als der des entgegengesetzten Magnetfeldes.
Daraus ergibt sich eine höhere Wahrscheinlichkeit für α-Spins, sodass es immer einen geringen Teil mehr an α-Spins als β-Spins in einem Gemisch gibt.
Entgegengerichtete Spins heben sich gegenseitig auf, was zur Folge hat, dass weniger Kerne existieren, die sich im energetisch günstigeren Zustand befinden. Diese übrig gebliebenen α-Spins sind für die Entstehung des NMR-Signals relevant. 9 10 11
Präzessionsfrequenz
Die variierenden Anordnungen der Kerne in einem Magnetfeld besitzen unterschiedliche energetische Zustände. Um von einem energieärmeren in ein energiereicheren Zustand überzugehen, muss dem Kern die exakte Energiedifferenz zwischen den beiden Energiezuständen zugeführt werden, d.h. der Kern muss mit Strahlung einer definierten Frequenz bestrahlt werden. Diese Frequenz wird Präzessionsfrequenz genannt und kann mit der Larmor-Gleichung ermittelt werden.
Wird mit der benötigten Frequenz bestrahlt, kommt es zu einer Resonanz (Spinumkehr). Der entsprechende Energiebetrag wird absorbiert und gemessen. Nachdem die Kerne angeregt wurden, fallen diese in den energieärmeren Zustand zurück und geben die absorbierte Energie als Wärmeenergie ab. Diese Rückkehr wird als Relaxation bezeichnet. Die zur Kernresonanz benötigte Energiemenge ist abhängig von der Umgebung der Kerne und drückt sich in der chemischen Verschiebung (Kapitel 1.5.1) aus. 12 13
Entstehung des NMR-Signals
Die Kernspins können als Vektoren zerlegt werden und die übrig gebliebenen Spins (siehe 1.3.2. Präzessionsbewegung) können anschließend zu einem Feldvektor addiert werden. Hieraus resultiert ein Feldvektor, der dem angelegten Magnetfeld gleichgerichtet ist. Dieser ist für die Entstehung des NMR-Signals relevant. Die Kerne werden mit hochfrequenten Radiowellen im 90° Winkel zum äußeren Magnetfeld bestrahlt (siehe Abb. 6). Es kommt zur Resonanz, wenn die Frequenz der eingestrahlten Radiowellen, der Präzessionsfrequenz entspricht.
⚠ $$ v_s=v_0=y/2π⋅B_0 ⚠ $$
⚠ $v_s$ =Einstrahlfrequenz [Hz]
⚠ $v_0$ =Frequenz des Atomkerns [Hz]
⚠ $B_0$ =Stärke des äußeren Magnetfelds [T]
Dies führt zur Quermagnetisierung/ transversaler Magnetisierung, d.h. die magnetischen Momente aller Kerne ändern sich. Der Feldvektor dreht sich transversal zum äußeren Magnetfeld, also um 90° in Richtung der Empfängerspule. Der Feldvektor lenkt aus, schneidet die Empfängerspule und nach kurzer Zeit schwenkt der Feldvektor zurück in Richtung des äußeren Magnetfeldes. Dieser Vorgang geschieht periodisch. Dadurch wird eine Spannung induziert und ein Kernresonanzsignal (Kerninduktion) kann gemessen werden.14
Messmethoden
Zur Kernresonanzmessung gibt es 2 Methoden: das Continous-wave-Verfahren (CW-Verfahren) und die Puls-Fourier-Transformations-NMR-Spektroskopie (PFT-NMR-Spektrometrie).
Continous-wave-Verfahren (CW-Verfahren)
Das Continous-wave-Verfahren beschreibt ein Verfahren, bei dem eine physikalische Größe kontinuierlich verändert wird. Nach der Lamor-Gleichung gibt es 2 Möglichkeiten zur Resonanzerzeugung (vs=v0). Beim sogenannten "Frequenz-Sweep-Verfahren" wird die Senderfrequenz vs bei konstanter Magnetfeldstärke B0 variiert. Beim sogenannten "Field-Sweep-Verfahren" wird die Magnetfeldstärke B0 bei konstanter Senderfrequenz vs verändert. Im Resonanzfall wird die verändernde Magnetisierung der Probe durch die Empfängerspule registriert. Diese Methode wird nur sehr selten verwendet, da sie eine geringe Empfindlichkeit aufweist und zudem sehr zeitaufwendig ist.15 16 17
Puls-Fourier-Transformations-NMR Spektroskopie (PFT-NMR-Spektrometrie)
Bei der PFT-NMR-Spektrometrie werden alle Atomkerne einer Kernsorte gleichzeitig durch einen sehr starken elektromagnetischen Impuls angeregt, sodass es zum Induktionsabfall kommt. Durch den Induktionsabfall entsteht eine Abklingkurve. Der gemessene abnehmende Strom in der Empfängerspule wird als free induction decay (FID) bezeichnet, und deren Abklingkurven werden zusammen addiert. Mithilfe der Fourier-Transformation (EDV) wird das FID in das NMR-Signal umgerechnet. Das Signal wird dabei mittels mathematischer Operationen mithilfe der Software von der Zeitdomäne (Intensität-Zeit-Signal) in die Frequenzdomäne (Intensität-Frequenz-Spektrum) umgewandelt, sodass man ein auswertbares Signal erhält. Die PFT-NMR-Spektroskopie ist viel empfindlicher, als das CW-Verfahren, da mehrere Spektren innerhalb einer kurzen Zeit aufgenommen und addiert werden können.18 19 20
1H-NMR-Spektroskopie
Signalverschiebung
Bei einer ersten Überlegung, wie ein NMR-Signal für jedes Element in der Theorie aussehen würde, käme man vermutlich auf die Idee, dass pro Element ein Peak im Diagramm entstehen müsse. Jedoch ist dies nur bedingt der Fall, da die Lage der Atome im Diagramm stark von ihren benachbarten Atomen abhängt. Dies wird als chemische Verschiebung δ bezeichnet.21
Auf der Skala dient als Nullpunkt die chemische Verschiebung von Tetramethylsilan ((CH3)4Si). Da es sich hierbei um eine sehr träge Substanz handelt, ist Tetramethylsilan ideal als Ausgangspunkt für die Bestimmung der Verschiebung von reaktiveren Substanzen geeignet.
Die chemische Verschiebung kann mit folgender Formel berechnet werden:
⚠ $$ \delta[ppm] = \frac{v(Probe)[Hz]-v(TMS)[Hz]×10^6}{Spektrometerfrequenz[Hz]} ⚠ $$
Ist ein Wert ermittelt worden, kann dieser auf der Skala entweder im hohen ppm-Bereich liegen, was als Tieffeldverschiebung aufgrund von Entschirmung passiert oder im niedrigen ppm-Bereich, was aufgrund von Hochfeldverschiebung durch Abschirmung geschieht. Um dies nachzuvollziehen, muss man sich die Larmor-Gleichung in Erinnerung rufen und folgendes beachten:
Die Resonanzfrequenz ist proportional zur Flussdichte des äußeren Magnetfeldes. Jedoch ist das Magnetfeld für jedes betrachtete Proton nicht gleich, sondern hängt von der Anwesenheit von Elektronen anderer Atome in direkter Nähe ab. Diese Elektronen manipulieren die Flussdichte so sehr, dass von einer effektiven Flussdichte die Rede ist, einer beeinflussten Flussdichte durch Elektronenpräsenz. Mithilfe dieser Erkenntnis sind die Begriffe Entschirmung und Abschirmung nun leicht zu erklären. Bei Entschirmung steigt die effektive Flussdichte, da hier elektronenziehende Atomgruppen in der Umgebung sind, was die Elektronendichte senkt und den Ausschlag im Diagramm nach links verschiebt, also ins tiefe Feld. Bei Abschirmung ist das Gegenteil der Fall. Hier sind elektronenschiebende Atomgruppen in der Umgebung, was die Elektronendichte erhöht und somit den Ausschlag nach rechts ins hohe Feld verschiebt.22
Signalintegral
Bei der Analyse eines NMR-Spektrums im Hinblick auf Protonen kann der Vergleich der Flächen unterhalb der Peaks als hilfreiche Methode herangezogen werden. Diese Flächen, auch Integrale genannt, geben das Verhältnis zwischen den verschiedenen Arten an Protonen im Molekül an. Jedoch muss darauf geachtet werden, dass die ermittelten Verhältnisse nicht zwangsläufig im analysierten Molekül in derselben Menge auftreten müssen. Einzig das Verhältnis der Mengen wird hiermit bestimmt und keine Mengenangaben der verschiedenen Protonenarten. Erstellt man eine Kurve der Integrale, kann man nun durch Vergleichen der Höhen der Intensitäten auf die Verhältnisse der Protonen schließen. Diese Kurve ist in NMR-Spektren oft oberhalb der Peaks angeordnet für einen übersichtlichen Vergleich der Integrale. In der folgenden Abbildung wurde schematisch ein Signalintegral für drei verschiedene Peaks dargestellt:

Bei genauerer Betrachtung fällt auf, dass das I. Integral eine Höhe von 3 Längeneinheiten (LE), das II. Integral eine Höhe von 2 LE und das III. Integral eine Höhe von 1 LE aufweist. Das Verhältnis zwischen den Signalen ist somit 3:2:1. Anhand dieses Verhältnisses, sowie einer Zuordnung der Signale zu den jeweiligen Arten von Protonen, kann man Informationen zur Strukturformel des untersuchten Moleküls ermitteln.
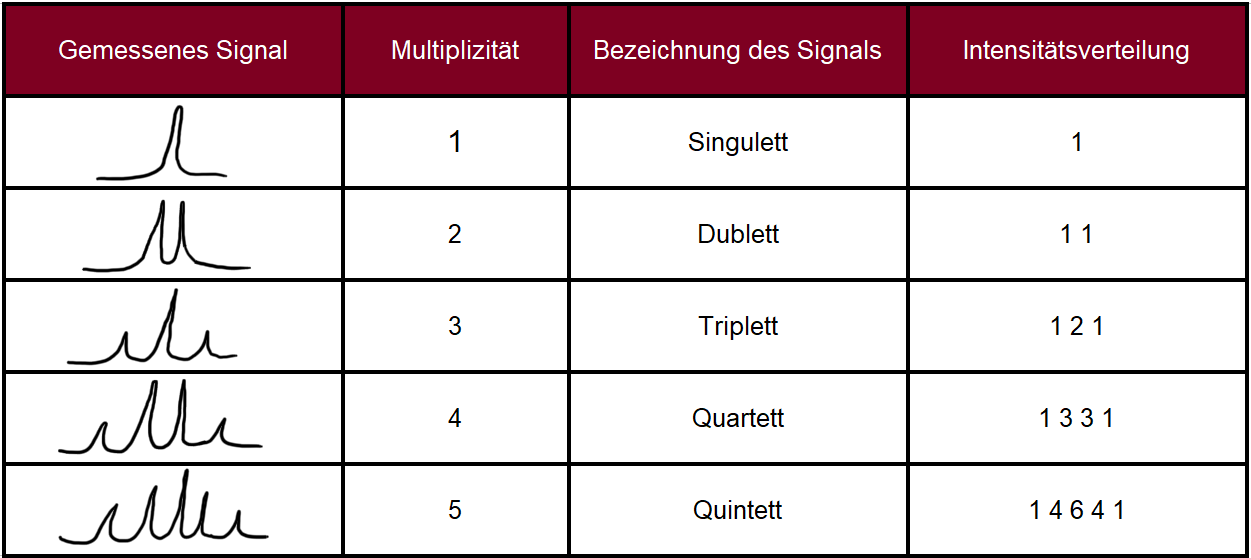
Spin-Spin-Kopplung
Bei genauerer Betrachtung verschiedener NMR-Diagramme fällt schnell auf, das manche Signale nicht nur aus einem Peak, sondern aus mehreren, dicht beieinander liegenden Peaks bestehen können. Um dies zu verstehen, ist die sogenannte "Spin-Spin-Kopplung" zu betrachten. Diese besagt, dass sich ein Signal in multiple Signale aufspalten kann. Dies geschieht durch eine Kopplung zwischen den Kernen in direkter Nähe, wobei zu berücksichtigen ist, dass nur Wasserstoffkerne hierzu in der Lage sind.24 Das Muster des aufgespaltenen Signals ist bei der Analyse der Kopplung besonders wichtig. Die folgende Tabelle liefert einen Überblick über mögliche Signalaufspaltungen:
Um den Ablauf der Spin-Spin-Kopplung genauer zu erklären, folgt nun ein Beispiel anhand von 1,1,2-Trichlorethan:26
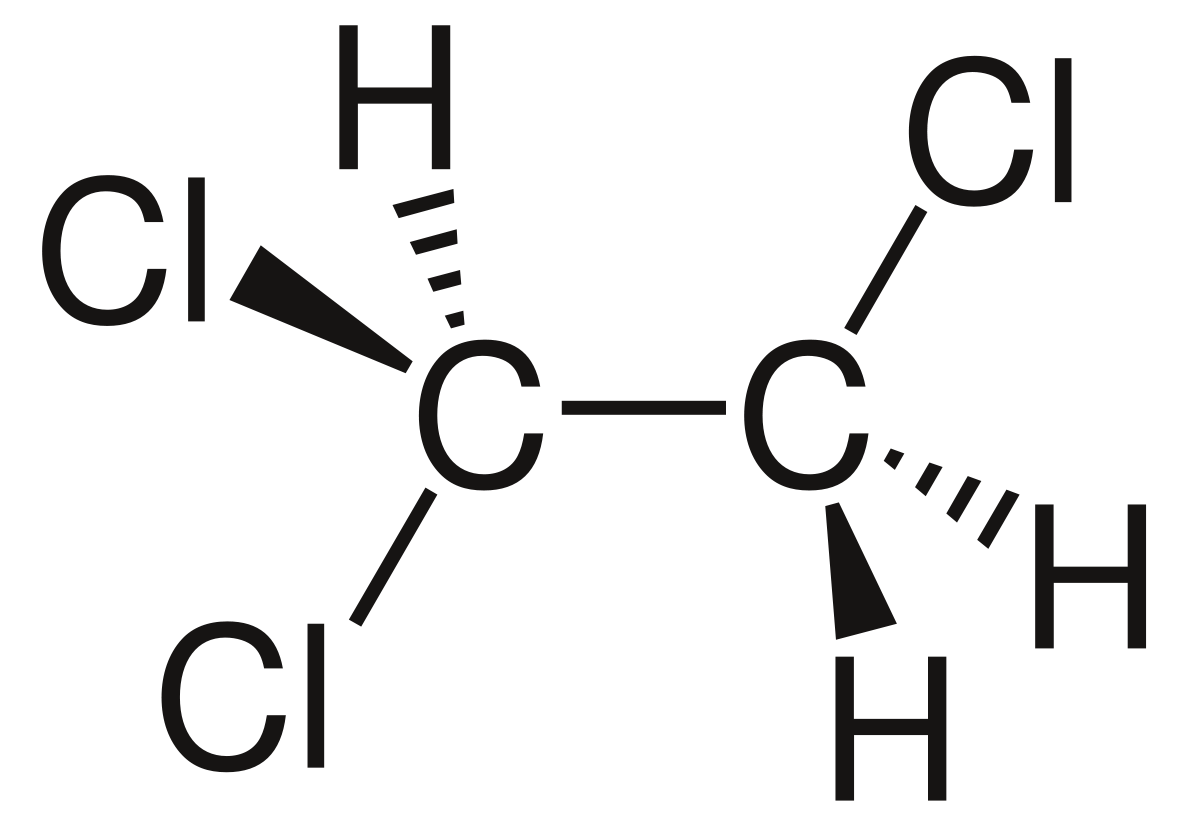
Bei Betrachtung des Moleküls ist erkennbar, dass insgesamt drei H-Atome vorliegen: Eins am ersten C-Atom und zwei am zweiten C-Atom. Vereinfacht werden diese folgend als CH und CH2 bezeichnet. Wichtig zu beachten ist zum einen, dass CH und CH2 benachbart sind, was die Spin-Spin-Kopplung erst ermöglicht, und zum anderen, dass ein Proton entweder einen Kernspin nach oben↑ oder unten↓ aufweisen kann. Dies hat zur Folge, dass eine Ausrichtung parallel und eine antiparallel zum äußeren Magnetfeld vorliegt.
Als nächstes wird das alleinige Proton von CH betrachtet und lässt es mit den Protonen von CH2 koppeln. Zuerst betrachtet man die möglichen Spin-Konfigurationen der Protonen von CH2. Diese können (oben-oben)↑↑, (oben-unten)↑↓, (unten-oben)↓↑ oder (unten-unten)↓↓ sein. Die Wahrscheinlichkeit für jede Konfiguration ist identisch, also 25 %. Die beiden gemischten Konfigurationen werden jedoch zu 50 % addiert, da sie zur selben Konfigurationsart gehören. Die Konfigurationstypen sehen also wie folgt aus: Typ 1: ↑↑ ; Typ 2: ↑↓ + ↓↑ ; Typ 3: ↓↓. Nun wird das Proton von CH mit jedem Konfigurationstyp der Protonen von CH2 einzeln gekoppelt, beginnend mit Typ 1. Zusätzlich wird festgelegt, dass das äußere Magnetfeld nach oben zeigt.
Bei Typ 1 (↑↑) zeigen beide Spins nach oben, also parallel zum äußeren Magnetfeld. Dies führt zu einer Erhöhung der effektiven Flussdichte für das Proton von CH, was zur Folge hat, dass sich die Frequenz und somit die chemische Verschiebung erhöht. Schlussendlich wird das Signal vom Proton im Diagramm in das tiefe Feld verschoben.
Bei Typ 2 (↑↓ + ↓↑) liegen die gemischten Spin-Ausrichtungen vor. Da sich diese neutralisieren, sorgen sie weder für Abschirmung noch Entschirmung des Signals unseres Protons, sodass es sich nicht verschiebt. Da Typ 2 jedoch eine Wahrscheinlichkeit von 50 % aufweist, ist der Peak doppelt so hoch wie der von Typ 1 oder 3, welche jeweils nur eine Wahrscheinlichkeit von 25 % besitzen.
Bei Typ 3 (↓↓) ist das exakte Gegenteil von Typ 1 der Fall. Die Spins zeigen beide entgegengesetzt zum Magnetfeld, sodass das effektive Magnetfeld des Proton von CH gesenkt wird. Folglich senkt sich die Frequenz, sodass das Signal in das hohe Feld verschoben wird. Fokussiert man sich wieder auf die Tabelle mit den möglichen Signalaufspaltungen, kann man die beschriebenen Signale in der dritten Zeile als ein sogenanntes Triplett wiederfinden.
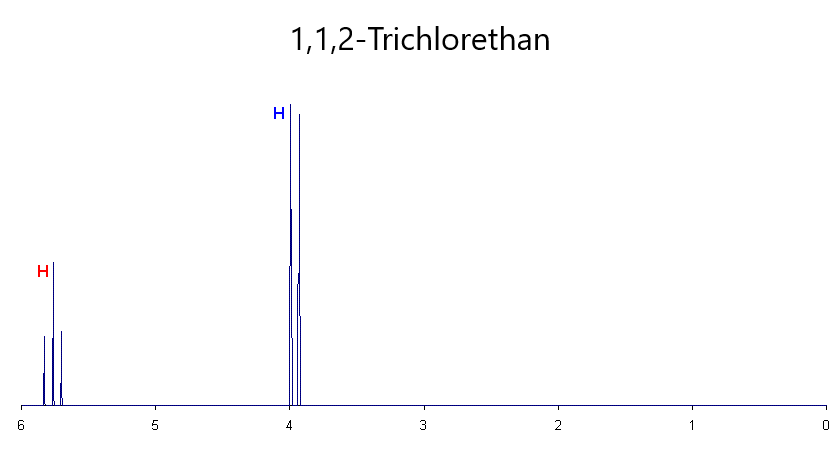
Zur Vollendung der Analyse von 1,1,2-Trichlorethan wird die Kopplung der Protonen von CH2 mit dem Proton von CH betrachtet. Hier gibt es am CH-Proton nur zwei Kernspinvarianten, entweder parallel oder antiparallel zum äußeren Magnetfeld. Daher können hier nur zwei Peaks im Diagramm entstehen: Zum einen das nach links verschobene Signal des parallel ausgerichteten Protons und das nach rechts verschobene Signal des antiparallel ausgerichteten Protons. Da jedoch die Kopplung von zwei Protonen betrachtet wird, sind die beiden Peaks dementsprechend doppelt so hoch im Vergleich zum Triplett. Dieses beschriebene Signal ist in der Abbildung 3 als Dublett wiederzufinden. Das gesamte NMR-Spektrum von 1,1,2-Trichlorethan ist in folgender Abbildung zur Verdeutlichung zu sehen:
Lösungsmittel
Eine weitere Komponente für das Verstehen eines NMR-Diagramms ist die Kenntnis über das verwendete Lösungsmittel. Grundsätzlich wird jedes Lösungsmittel, welches Wasserstoff beinhaltet, vor Gebrauch deuteriert. Hierbei werden die enthaltenen Wasserstoffkerne durch Deuteriumkerne ersetzt. Chemisch gesehen ist Deuterium nahezu identisch mit Wasserstoff, jedoch besitzt es ein Neutron im Kern und weist daher das doppelte molare Gewicht auf. Da die Kernmassen von Deuterium und Wasserstoff abweichen, sie jedoch dieselbe Anzahl an Protonen und Elektronen besitzen, handelt es sich bei Deuterium um ein Isotop von Wasserstoff. Eine problematische Tatsache bei der Deuterierung von Lösungsmittel ist, dass sie nie vollständig abläuft. Dies bedeutet, dass noch Wasserstoffkerne und Wassermoleküle im Lösungsmittel vorhanden sind, welche jeweils für getrennte Störsignale im Diagramm sorgen. Ist man sich jedoch dessen bewusst, kann man diese Signale im Diagramm ausblenden und als Störungen abhaken. Für einige Lösungsmittel gibt es hierfür tabellarische ppm-Werte zur Kontrolle.
Ein besonderes Lösungsmittel ist das Deuteriumoxid (D2O), welches auch als „Schweres Wasser“ bezeichnet wird, da beide Wasserstoffatome im Molekül durch doppelt so schwer wiegende Deuteriumkerne ausgetauscht wurden. Dieses Lösungsmittel bringt den Vorteil mit sich, Protonen von leicht deprotonierbaren Atomgruppen, wie beispielsweise einer OH-Gruppe, durch ein Deuteriumkern ersetzen zu können. Es erfolgt somit ein Austausch der Kerne, sodass aus dem D2O ein HOD und aus der OH-Gruppe eine OD-Gruppe entsteht. Dieser Vorgang wird als "H/D-Tausch" bezeichnet. Da die entstandene OD-Gruppe erzeugt ein Signal bei ca. 4 ppm. Somit eignet sich der "H/D-Tausch" ideal dafür, unbekannte Substanzen, die mittels der NMR-Spektroskopie identifiziert werden sollen, auf genau diese chemischen Gruppen mit leicht abspaltbarem Wasserstoff zu überprüfen.29
13C-NMR-Spektroskopie
Prinzip
Das Kohlenstoffisotop 13C zeigt eine Kernspinquantenzahl I = 0,5. Dadurch ist es zugänglich für Kernresonanzmessungen. Prinzipiell gelten somit die selben Gesetzmäßigkeiten wie bei der 1H-NMR-Spektroskopie. Jedoch hat die 13C-NMR-Spektroskopie eine geringere Empfindlichkeit als die 1H-Spektroskopie.
Die natürliche Häufigkeit des 13C-Isotops liegt nur bei ca. 1,1% und das gyromagnetische Verhältnis von 13C ist nur ungefähr ¼ so groß wie das von 1H (die 13C-Kernmagnete sind nur etwa ein Viertel so stark wie die 1H-Kernmagnete).30 Deshalb musste die Empfindlichkeit der Messungen durch die Einführung der Puls-Fourier-Transformations-NMR-Spektroskopie(PFT-NMR-Spektroskopie, siehe Kapitel 1.4.2), verbessert werden, um 13C-Messungen durchführen zu können.31
Chemische Verschiebung der 13C-Atome
Im Vergleich zu Wasserstoffkernen ist der Bereich der chemischen Verschiebung bei 13C-Kernen wesentlich größer. Der spektrale Bereich erstreckt sich von 0 bis 220 ppm. Die chemische Verschiebung ist abhängig von dem Hybridisierungsgrad des Kohlenstoffatoms, Substituenteneinflüssen und der Elektronendichte.32
Spin-Kopplungen
In der 13C-NMR-Spektroskopie treten Homonukleare Kopplungen (13C-13C-Kopplungen) und Heteronukleare Kopplungen (z.B. mit 1H, 19F-Kernen) auf.
Die homonuklearen Kopplungen sich durch die geringe Häufigkeit des 13C-Isotops zu vernachlässigen.
Bei den heteronuklearen Kopplungen ist die Kopplung mit 1H-Kernen zu nennen. Da sowohl 1H als auch 13C die Kernspinquantenzahl I= 0,5 aufweisen, gelten bei 1H-13C-Kopplungen die gleichen Multiplizitätsregeln wie in der 1H-NMR-Spektroskopie. Kohlenstoffatome von CH3-Gruppen bilden daher ein Quartett, von CH2-Gruppen ein Triplett und von CH-Gruppen ein Dublett. Quartäre C-Atome liegen als Singuletts vor.33
Entkopplungsverfahren in der 13C-NMR-Spektroskopie
Durch die großen 1H/13C-Kopplungskonstanten kommt es zu einer Überlagerung von Multipletts.34 Deshalb sind Entkopplungsverfahren in der 13C-NMR-Spektroskopie von großer Bedeutung. Für die Entkopplung gibt es verschiedene Methoden:
Protonen-Breitband-Entkopplung
Bei der Protonen-Breitband-Entkopplung werden alle 13C-1H-Kopplungen aufgehoben und die Signalintensität wird erhöht.35
Protonen-Off-Resonance-Entkopplung
Dieses Verfahren hat die Vorteile, dass die 13C-1H-Kopplungen erhalten bleiben und die Kopplungskonstanten verkleinert werden.36
Gepulste Protonen-Entkopplung
Diese Methode gestattet die Unterscheidung wasserstofftragender von nicht an Wassserstoff gebundenen Kohlenstoffatome. Signale für CH-, CH2-, CH3-Gruppen werden erhöht und Signale für quartäre Kohlenstoffatome annähernd bleiben gleich.37
Selektive 1H-Entkopplung
Sind die Resonanzfrequenzen der Protonen aus dem 1H-NMR-Spektrum bekannt, können C-H-Bindungen eindeutig zugeordnet werden.38
Instrumenteller Aufbau
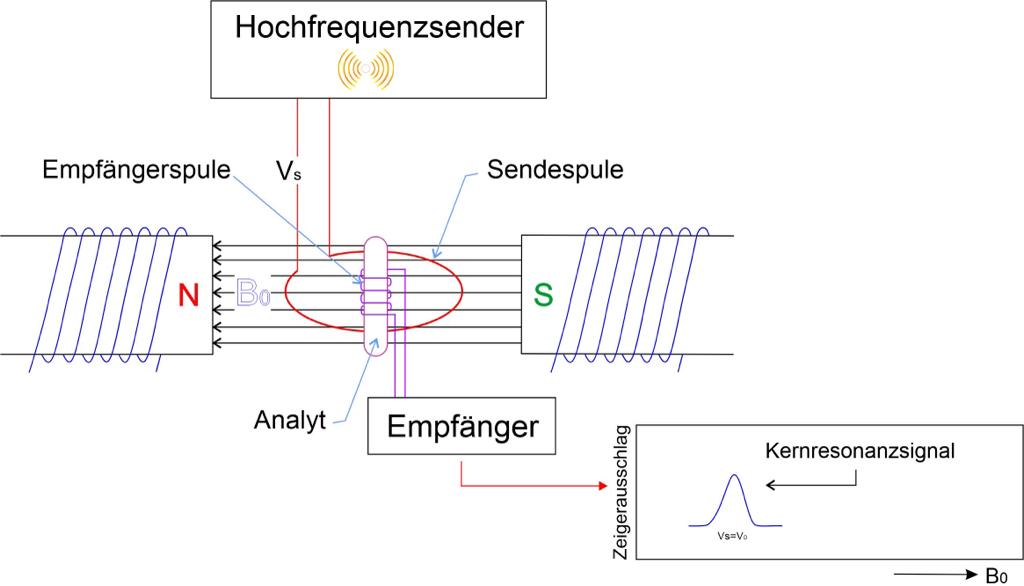
Das von Felix Bloch entwickelte Kernresonanzspektrometer hat folgende Bestandteile:
-Homogenes Magnetfeld der Stärke B0, in welches die Probe eingebracht wird (Elektromagnet oder Permanentmagnet)
-Hochfrequenzsender zur Bestrahlung der Probe
-Messeinrichtung (Empfänger), die den Betrag der von den Kernen aufgenommenen und wieder abgegebenen Energie misst und im Kernresonanzspektrum registriert.
Der Aufbau kann eines Kernresonanzspektrometers lässt sich in dem Bild gut erkennen. Der Analyt befindet sich in einem Glasröhrchen, welches von einer Empfängerspule (pinke Spule im Bild) umgeben ist. Das Glasröhrchen rotiert um seine Längsachse, sodass horizontale Feldinhomogenitäten vermieden werden. Von der Sendespule (Vs, rote Spule) wirkt senkrecht zur Magnetfeldrichtung B0 die elektromagnetische Strahlung des Hochfrequenzsenders auf die Probe ein. Die Empfängerspule steht senkrecht zur Sendespule und zum Magnetfeld. Der Empfänger ist mit einem Datenverarbeitungssystem gekoppelt.40
Moderne NMR-Geräte besitzen einen spulenförmigen Kryomagneten, welcher aus supraleitenden Seltenen-Erde-Legierungen besteht. Da diese Magnetspule stets gekühlt werden muss, befinden sich in dem doppelwandigen Dewargefäß flüssiges Helium und flüssiger Stickstoff. Nach einmaliger Aufladung der Spule kommt es zu einem dauerhaften Stromfluss, da durch die Kühlung kein Widerstand mehr vorhanden ist.41
Einzelnachweise
1 Skript zum Seminar von Dr. Oliver Orban: NMR – 1, Sommersemester 2021: Einführung und Grundlagen der NMR-Spektrometrie, Seite 3-6 ⇑
2 Dominik, Steinhilber, Wurglics; Instrumentelle Analytik kompakt; 3.Auflage; Stuttgart: Wissenschaftliche Verlagsgesellschaft, 2013; Seite 128 f. ⇑
3 https://www.analytik.ethz.ch/vorlesungen/biopharm/Spektroskopie/NMR.pdf; [letzter Zugriff: 31.05.2021, 13:33 Uhr] ⇑
4 Skript zum Seminar von Dr. Oliver Orban: NMR – 1, Sommersemester 2021: Einführung und Grundlagen der NMR-Spektrometrie, Seite 7 ff. ⇑
5 Dominik, Steinhilber, Wurglics; Instrumentelle Analytik kompakt; 3.Auflage; Stuttgart: Wissenschaftliche Verlagsgesellschaft, 2013 ; Seite 128 f. ⇑
6 https://www.analytik.ethz.ch/vorlesungen/biopharm/Spektroskopie/NMR.pdf; [letzter Zugriff: 31.05.2021, 13:33 Uhr] ⇑
7 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite 240-244 ⇑
8 eigens erstellte Tabelle 1: in Anlehnung an Dr. Oliver Orban, Skript NMR – 1, Sommersemester 2021: Seite 9-12 ⇑
9 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite: 240-244 ⇑
10 Skript zum Seminar von Dr. Oliver Orban: NMR – 1, Sommersemester 2021: Einführung und Grundlagen der NMR-Spektrometrie, Seite 11-17 ⇑
11 Dominik, Steinhilber, Wurglics; Instrumentelle Analytik kompakt; 3.Auflage; Stuttgart: Wissenschaftliche Verlagsgesellschaft, 2013; Seite 128 f. ⇑
12 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite: 240-244 ⇑
13 https://www.uni-muenster.de/imperia/md/content/pharmaz_und_med_chemie/studieren/semester/8semester/nmrtischvorlage.pdf [letzter Zugriff: 20.06.2021, 13:02 Uhr] ⇑
14 Skript zum Seminar von Dr. Oliver Orban: NMR – 1, Sommersemester 2021: Theoretischer Hintergrund, Seite 15 ff. ; NMR-Signal, Seite 18-21] [^Dominik, Steinhilber, Wurglics; Instrumentelle Analytik kompakt; Seite 128 f.; 3.Auflage; Stuttgart: Wissenschaftliche Verlagsgesellschaft, 2013 ⇑
15 Skript zum Seminar von Dr. Oliver Orban: NMR – 1, Sommersemester 2021: Messmethoden, Seite 22 f. ⇑
16 Dominik, Steinhilber, Wurglics; Instrumentelle Analytik kompakt; 3.Auflage; Stuttgart: Wissenschaftliche Verlagsgesellschaft, 2013; Seite 128 f. ⇑
17 https://www.chemie.uni-kl.de/fachrichtungen/oc/kubik/index.php?lan=de&lev1=0tea&lev2=oc3&lev3=nmr [letzter Zugriff: 31.05.2021, 17:33 Uhr] ⇑
18 Skript zum Seminar von Dr. Oliver Orban: NMR – 1, Sommersemester 2021: Messmethoden, Seite 22 f. ⇑
19 Dominik, Steinhilber, Wurglics; Instrumentelle Analytik kompakt; 3.Auflage; Stuttgart: Wissenschaftliche Verlagsgesellschaft, 2013; Seite 128 f. ⇑
20 https://www.chemie.uni-kl.de/fachrichtungen/oc/kubik/index.php?lan=de&lev1=0tea&lev2=oc3&lev3=nmr [letzter Zugriff: 31.05.2021, 17:33 Uhr] ⇑
21 Skript zum Seminar von Dr. Oliver Orban: NMR – 2, Sommersemester 2021: Chemische Verschiebung, Seite 4 ⇑
22 Skript zum Seminar von Dr. Oliver Orban: NMR – 2, Sommersemester 2021: Chemische Verschiebung, Seite 7 ff. ⇑
23 eigens erstellte Abbildung ⇑
24 Skript zum Seminar von Dr. Oliver Orban: NMR – 2, Sommersemester 2021: Spin-Spin-Kopplung, Seite 17 ⇑
25 eigens erstellte Tabelle ⇑
26 https://www.youtube.com/watch?v=QlM21sl-TDg [letzter Zugriff: 31.05.2021, 21:11 Uhr] - (Minute 6:25 - 12:21) ⇑
27 https://de.wikipedia.org/wiki/1,1,2-Trichlorethan#/media/Datei:1,1,2-Trichlorethan(Punkt)svg - [letzter Zugriff: 17.06.2021, 02:49 Uhr] ⇑
28 https://upload.wikimedia.org/wikipedia/commons/1/15/1H_NMR_C02H03Cl3_1%2C1%2C2-trichloorethaan(Punkt)png - [letzter Zugriff: 20.06.2021, 20:19 Uhr] - (im Bild wurde die Strukturformel entfernt und die Überschrift ersetzt) ⇑
29 Skript zum Seminar von Dr. Oliver Orban: NMR – 2, Sommersemester 2021: Lösemittel/ H/D-Tausch, Seite 31 f. ⇑
30 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite 293 ⇑
31 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite 293 ⇑
32 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite 293 ⇑
33 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite 307 ⇑
34 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite 310 ⇑
35 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite 310 ⇑
36 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite 310 ⇑
37 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite 310 ⇑
38 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite 310 ⇑
39 eigens erstellte Abbildung ⇑
40 Rücker, G., Neugebauer, M.& Willems, G. G. (2013): Instrumentelle Pharmazeutische Analytik. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft Stuttgart. Seite 247 ⇑
41 Skript zum Seminar von Dr. Oliver Orban: NMR – 1, Sommersemester 2021: Das NMR-Gerät, Seite 24 ⇑
Monographiebeispiel: Heparin-Natrium, Prüfung auf Identität
Stoffcharakterisierung
Heparin-Natrium gehört zu der Gruppe der Heparine und ist das Natriumsalz eines sulfatierten Glycosaminoglycans. Dieses kommt in den Intestinalgeweben der Säugetiere vor und wird daher, für weitere Anwendungen, aus diesen Geweben isoliert. Wichtige Bausteine des Heparin-Natriums sind D-Glucosamin, D-Glucuronsäure, L-Iduronsäure, Essigsäure und Schwefelsäure, die aus der Hydrolyse des Heparin-Natriums resultieren. Heparin-Natrium zeigt eine gute Wasserlöslichkeit und ist ein weißes bis fast weißes, hygroskopisches Pulver. Zudem wirkt es entzündungshemmend, schmerzstillend sowie resorptionsfördernd. Darüber hinaus ist Heparin-Natrium in der Lage, die Blutgerinnung zu hemmen. Es ist also ein Koagulans. Diese Fähigkeit kommt dadurch zustande, dass Heparin-Natrium als Katalysator für die Inaktivierung der Gerinnungsfaktoren fungiert. Durch die katalysierte Reaktion werden die Gerinnungsfaktoren von Antithrombin in ihrer blutgerinnenden Funktion gehemmt. Somit findet Heparin-Natrium, sowohl prophylaktisch als auch therapeutisch, Verwendung bei Beschwerden wie Krampfadern, Blutergüssen oder Behandlungen, die durch die Blutgerinnung gestört werden könnten.1 2 3 4
Durchführung der Monographie
Die Untersuchung wird im Europäischen Arzneibuch nach Methode B. Kernresonanzspektroskopie 2.2.33 wie folgt durchgeführt:
Das Arzneibuch gibt drei Lösungen vor, die für die Durchführung der Identitätsprüfung vorbereitet werden sollen: Lösung A wird als Grundlage für die Untersuchungs- sowie Referenzlösung vorbereitet. Sie erfolgt durch Lösen des Natriumtrimethylsilyl-(D4)propionat R (C6H9D4NaO2Si) in (D2)Wasser R (20 µg · ml−1). Anschließend wird die Lösung im Spektrometer vermessen. Man vergleicht die Signale im 1H-NMR-Spektrum bei 5,22 ppm und 5,44 ppm miteinander. Das Signal bei 5,22 ppm sollte hierbei in der Länge nicht kleiner sein, als 80 % des Signals bei 5,44 ppm. Das heißt, dass das Signal demnach mindestens 1/5 der Größe haben sollte als das Signal bei 5,44 ppm. Sollte dies nicht zutreffen, dann versetzt man die Lösung mit Natriumedetat R (12 µg · ml−1) zur Streckung des Signals und überprüft erneut die Signale. Eine weitere Untersuchungslösung sowie eine entsprechende Referenzlösung werden im Anschluss aus der bereits vorbereiteten Lösung A hergestellt, indem man jeweils 0,7ml dieser Lösung mit 20 mg Substanz für die Untersuchungslösung und 20 mg Heparin-Natrium für die Referenzlösung vermengt. Diese werden dann mit einem Spektrometer mittels Fouriertransformationsverfahren (FT-NMR), mit einer eingestellten Frequenz von mindestens 300 MHz und bei einer Temperatur von 25°C nacheinander aufgenommen. Weitere Einstellungsfaktoren sind der spektrale Bereich, der sich zwischen 0 bis 10 – 12 ppm erstrecken soll; die Erfassungszeit befindet sich in einem Bereich zwischen zwei bis vier Sekunden und die Pulsbreite befindet sich zwischen einem Winkel von 30° - 90°. Es sollen mindestens 16 Messungen erfolgen. Das Signal-Rausch-Verhältnis beträgt mindestens 200/1 im Bereich nahe 2 ppm.5
Auswertung/ Interpretation/ Bedeutung und Eignung der analytischen Methode
Die aufgenommenen Spektren der zu untersuchenden Lösung sowie der Referenzlösung sollten sich in ihren Intensitäten so weit ähneln, dass man sie gut miteinander vergleichen kann. Intensitätsvariationen im Bereich zwischen 3,35 ppm und 4,55 ppm sind dennoch möglich. Signale in Form von eventuellen Verunreinigungen, die bei der Herstellung aufgetreten sein können und auch das Signal des Lösemittels werden ebenfalls aufgezeichnet und sollten identifiziert werden. Es können auch weitere noch nicht identifizierte Signale auftreten, die in ihrer Größe 4 % der Höhe, verglichen mit dem Heparinsignal ausmachen. Diese eventuell vorkommenden Signale dürfen nicht in den folgenden ppm-Bereiche auftreten: 0,10 – 2,00 ppm; 2,10 – 3,10 ppm; sowie 5,70 – 8,00 ppm. Bei 0,00 ppm ist das Referenzsignal Trimethylsilyl-(D4)propionat sichtbar. Ein mögliches Methylsignal bei ca. 2,08 ppm kann, mit einer evtl. Abweichung von +/- 0,02 ppm, vorliegen. Das wäre das entsprechende Signal für Dermatansulfat. Das Signal für die zu untersuchende Substanz Heparin wird im Spektrum als Signal bei 5,42 ppm sichtbar. Die intensivsten Signale der zu untersuchenden Substanz Heparin-Natrium liegen bei den folgenden ppm-Werten: 2,04 ppm, bei 3,27 ppm sollte ein Dublett sichtbar werden, bei 3,34 ppm, 5,22 ppm und 5,42 ppm. Die Abweichungen dürfen maximal +/- 0,03 ppm betragen. Für NMR-Spektren sind entsprechende Lösemittel vorhergesehen, damit eine entsprechende Signalzuordnung erfolgen kann. Lösemittel werden ebenfalls in Form von Signalen sichtbar. Dabei könnte es zu Signalüberlagerungen kommen, sodass die Signale der zu untersuchenden Substanz überdeckt werden und folglich die Auswertung erschwert wird. Im Arzneibuch wird (D2)-Wasser verwendet, da deuterierte Lösungsmittel im Spektrum praktisch keine bis geringe Signale auslösen und somit die Signalzuordnung erleichtern.6 7 Natriumtrimethylsilylpropionat wird als Referenzsubstanz in der Kernresonanzspektroskopie eingesetzt. Sie liegt im Spektrum bei 0,00 ppm, wo keine Überlappung mit anderen Signalen zu befürchten sind. Das liegt an einer anderen Resonanzfrequenz, verglichen mit einem Großteil der organischen Moleküle.8
Einzelnachweise
1 https://arzneibuch.de/arzneibuch/arzneibuch/start.xav?lang=de [letzter Zugriff: 30.05.2021, 10:37 Uhr] ⇑
2 https://www.pharmawiki.ch/wiki/index.php?wiki=Heparin-Natrium [letzter Zugriff: 30.05.2021, 10:49 Uhr] ⇑
3 https://www.ratiopharm.de/produkte/details/praeparate/praeparatedaten/detail/pzn-3029843.html [letzter Zugriff: 30.05.2021, 13:18 Uhr] ⇑
4 https://www.dna-diagnostik.hamburg/analysen/antithrombin-mangel-serpinc1/ [letzter Zugriff: 30.05.2021, 11:04 Uhr] ⇑
5 Ph.Eur., 2020; Bracher, F./Heisig, P./Langguth, P., 2020 ⇑
6 https://de.wikipedia.org/wiki/Deuterierte_Verbindung [letzter Zugriff: 31.05.2021, 21:33 Uhr] ⇑
7 http://www.akoci.uni-hannover.de/ak-duddeck/pdf/pdf-spektro-info/NMR-Loesungsmittel.pdf [letzter Zugriff: 31.05.2021, 21:37 Uhr] ⇑
8 https://de.wikipedia.org/wiki/Natriumtrimethylsilylpropionat [letzter Zugriff: 31.05.2021, 21:53 Uhr] ⇑
TU-Braunschweig Institut für Medizinische und Pharmazeutische Chemie Seminar: Instrumentelle Analytik Kontakt: tubs@t-kellner.de
