Karl-Fischer-Titration
Titelblatt
Bericht der Expertengruppe für
Karl-Fischer-Titration
SoSe 2021
Abgabedatum
21.06.2021
Über-/Expertengruppe 01
Nadja Drischel
Emina Osmic
Violetta Safraider
Ira Sharon Senftleben
Tiana Wargers
Mark Wellmann
Karl-Fischer-Methode
Inhaltsverzeichnis
Einleitung
Die Karl-Fischer-Titration (kurz KF) ist ein maßanalytisches Verfahren zur Bestimmung des Wassergehaltes einer flüssigen oder festen Probe, welches von Dr. Karl Fischer erstmalig zum Zwecke der Wasserbestimmung von Schwefeldioxid beschrieben wurde. Sie dient als Standardmethode, da alternative Methoden wie die thermogravimetrische Verfahren, beispielsweise der Trocknungsverlust, Kristallwasser nicht restlos entfernen und leicht flüchtige Probenbestandteile miterfassen. 1 2 Mit ihr wird aus pharmazeutischer Sicht der Wassergehalt von einigen organischen Lösungsmitteln, aber auch von ätherischen Ölen, Salben oder anderen organischen Substanzen ermittelt. 3
Grundlagen
Chemische Grundlagen
Reaktionen
Als Grundlage der KF dient die Bunsen-Reaktion, welche die Oxidation von Schwefeldioxid mit Iod darstellt und nur unter der Anwesenheit von Wasser abläuft. 4 Es werden zur Bildung von der Schwefelsäure zwei Mol Wasser pro Mol Iod benötigt.

Der Zusatz einer Base (hier Pyridin) ist zwingend notwendig, um die entstehenden Protonen abzupuffern. Dies verschiebt das Gleichgewicht auf die Produktseite. Weitere mögliche Basen sind Imidazol, 2-Methylaminopyridin, Diethanolamin, Acetat, Dichloracetat, Salicylat u. a.. Darüber hinaus erfüllt die Base einen weiteren Zweck: So verhindert Pyridin das Austreten von SO2 aus Lösungen, indem es ein schwerflüchtiges Addukt (SO2 ⋅ Base) bildet und daher den Dampfdruck senkt. 6 Als Lösungsmittel wird oft Methanol verwendet, um auch polare Substanzen gut lösen zu können. Andere Alkohole sind auch einsetzbar. 7 Zu beachten ist, dass Methanol ebenfalls als Reaktionspartner beteiligt ist und einen Methylschwefligsäureester bildet, wodurch sich das stöchiometrische Verhältnis von Wasser zu Iod von 2:1 auf 1:1 ändert und damit die Empfindlichkeit erhöht. 8
Aufgrund der Toxizität substituiert Imidazol das Pyridin in modernen Reagenzlösungen. 9

Die Maßlösung dieser Redoxtitration stellt ein Gemisch aus Iod, der gewählten Base und dem Schwefeldioxid da, welches als Karl-Fischer-Reagenz bezeichnet wird.
Störungen
Das Störpotenzial der Karl-Fischer-Titration bezieht sich im Allgemeinen auf Substanzen, die unter Aufnahme oder Abgabe von Wasser dessen Gehalt ändern, sowie auf Substanzen, welche I2 reduzieren oder I- oxidieren und so einen anderen Wassergehalt vorspielen. 11
Diese Reduktions- und Oxidationsmittel sind vielfältig und reichen von Thiolen und Metallsalzen bis hin zu klassischeren Beispielen wie der Ascorbinsäure und Nitriten oder Sauerstoff und Permanganaten.
Methanol regiert unter Anwesenheit von Aldehyden und Ketonen unter Wasserbildung zu Halb- bzw. Vollacetalen. Dieses Wasser würde dann zusätzlich erfasst werden. Des Weiteren können Aldehyde auch in Form einer Bisulfitaddition Wasser verbrauchen. Bei nicht ausreichend gepufferten Lösungen besteht ebenfalls die Möglichkeit einer sauren Veresterung von vorhandenen Säuren mit Methanol, wobei ebenfalls Wasser entsteht. Einige Oxide, Hydroxide und Carbonate reagieren mit Protonen unter Wasserabspaltung, weshalb dann die Methode des Trocknungsverlustes vorgezogen wird. Beidem kann mit ausreichender Neutralisierung und Pufferung entgegengewirkt werden. 12
Physikalische Grundlagen
Biamperometrische Indikation
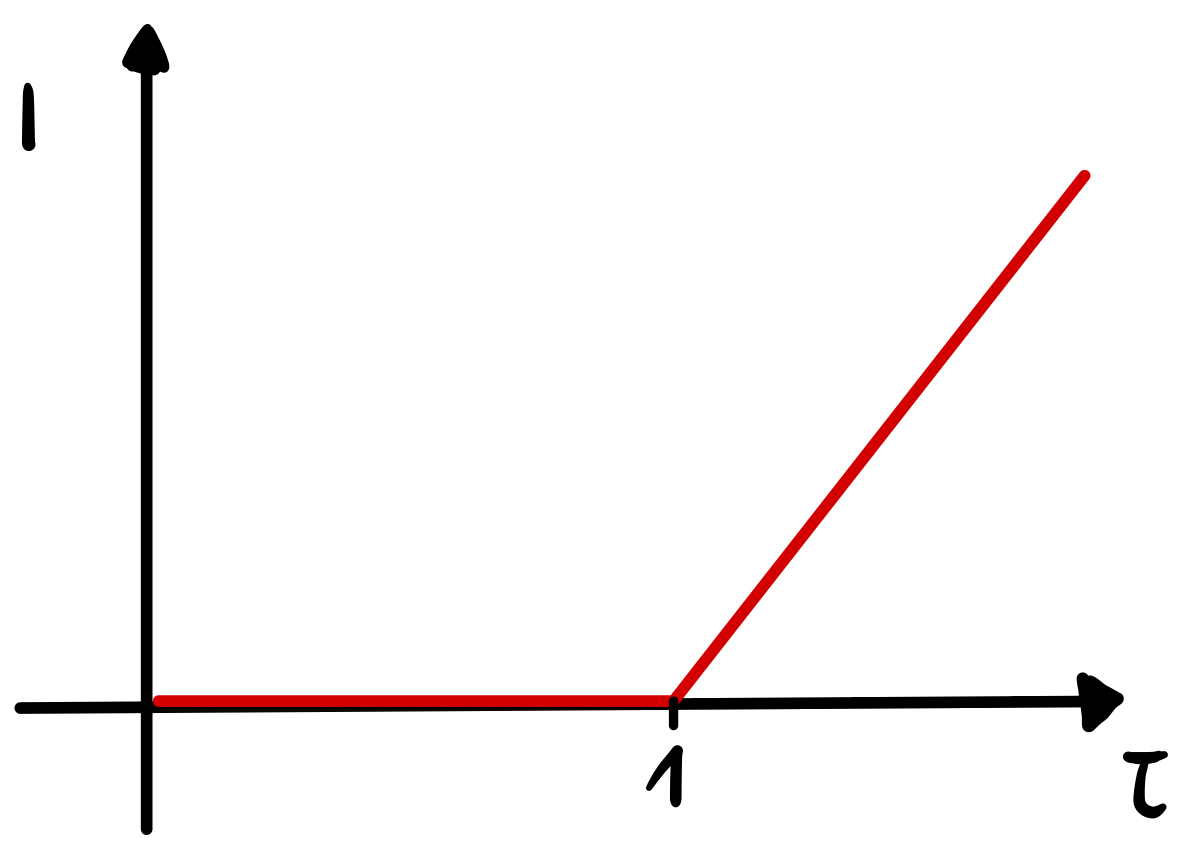
Die Biamperometrie ist eine voltammetrische Methode, die als elektrochemisches Indikationsverfahren genutzt werden kann. So kann der Endpunkt einer Titration angezeigt werden. 14 Voltammetrie ist ein Begriff, der sich aus Voltametrie und Amperometrie zusammensetzt und grundlegend auf Strom-Spannungs-Kurven beruht. 15 Bei der Biamperometrie wird bei einer konstant angelegten Polarisationsspannung die Stromstärke gemessen. Eingesetzt wird eine Doppelplatinelektrode als Messkette. Zwischen den zwei Platinelektroden wird ein Potential angelegt, das möglichst gering sein soll, damit nur eine Reaktion erfasst wird. Damit aber ein Strom fließen kann, müssen an den Elektroden Reaktionen stattfinden. Das anliegende Potenzial reicht aus, um Reaktionen reversibler Redoxsysteme auszulösen. An der Anode findet dann die Oxidation und an der Kathode die Reduktion statt. Fehlt ein Reaktionspartner, fließt kein Strom. 16
Nutzt man dieses Verfahren für die Endpunktsindikation der Karl-Fischer-Titration, muss eine höhere Polarisationsspannung (500 mV) als üblich angelegt werden, da im wasserfreien Medium gearbeitet wird und somit der Innenwiderstand der Lösung höher ist. Durch die Gültigkeit des Ohm’schen Gesetzes muss eine höhere Spannung angelegt werden, um einen Strom messen zu können. Der Endpunkt der Titration wird angezeigt durch eine sogenannte Kick-off-Kurve (siehe Abbildung). Vor dem Äquivalenzpunkt liegt kein reversibles Redoxsystem vor, weil das Iod des Karl-Fischer-Reagenzes direkt für die Umsetzung des Wassers verbraucht wird. Es fließt somit kein Strom. Nach dem Äquivalenzpunkt liegen sowohl Iod als auch Iodid frei in der Lösung vor, sodass ein reversibles Redoxsystem vorhanden ist. An der Anode findet nun die Oxidation von Iodid zu Iod und an der Kathode die Reduktion von Iod zu Iodid statt. Es kommt zu einem messbaren Stromfluss, sodass der Verbrauch an Maßlösung zu diesem Zeitpunkt abgelesen werden kann. 17
Bivoltametrische Indikation
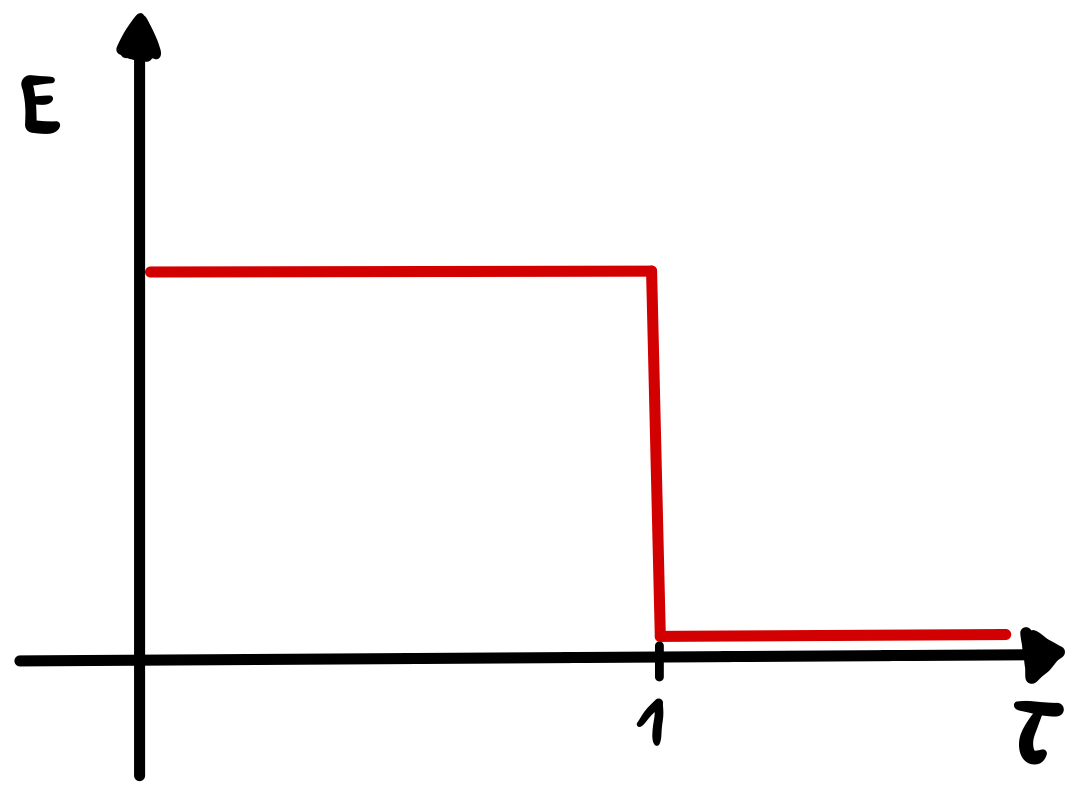
So wie die Biamperometrie ist auch die Bivoltametrie eine voltammetrische Methode, die als elektrochemisches Indikationsverfahren eingesetzt werden kann. Auch hier wird eine Doppelplatinelektrode als Messkette eingesetzt. Allerdings wird hier keine Spannung, sondern ein konstanter Polarisationsstrom an den beiden Platinelektroden angelegt, der weitestgehend gering gehalten werden soll, um die Messung möglichst wenig zu beeinflussen. Damit dieser Strom fließen kann, müssen an der Anode Oxidations- und an der Kathode Reduktionsreaktionen stattfinden. Dabei werden an den Elektroden lediglich die Stoffe oxidiert und reduziert, die die geringste Potentialdifferenz zwischen der Oxidation und der Reduktion aufweisen. Eine solch geringe Potentialdifferenz weisen reversible Redoxsysteme auf. 19
Wird diese Form der Endpunktsindikation bei der Karl-Fischer-Titration angewendet, muss ein höherer Polarisationsstrom (20 µA) angelegt werden als üblich, um ein Potential messen zu können, da im wasserfreien Medium gearbeitet wird und der Innenwiderstand somit höher ist. Der Endpunkt wird dadurch angezeigt, dass am Äquivalenzpunkt ein sehr geringes Potential von wenigen mV gemessen wird (siehe Abbildung), weil dann ein reversibles Redoxsystem aus freiem Iod des Karl-Fischer-Reagenzes und freiem Iodid, das durch die Reaktion mit dem Wasser entstanden ist, vorliegt. Vor dem Äquivalenzpunkt liegt kein reversibles Redoxsystem vor, weil das Iod als Maßlösung fungiert und das Wasser in der Probe umsetzt. Es werden andere Bestandteile an der Anode oxidiert und an der Kathode reduziert, die eine höhere Potentialdifferenz aufweisen und somit auch ein hohes Potential gemessen wird. Oftmals wird ein Grenzwert für das Potential gezogen. Wird dieser bei der Titration unterschritten, stoppt die Titration und der Verbrauch an Maßlösung kann abgelesen werden. 20
Potentiometrische Indikation
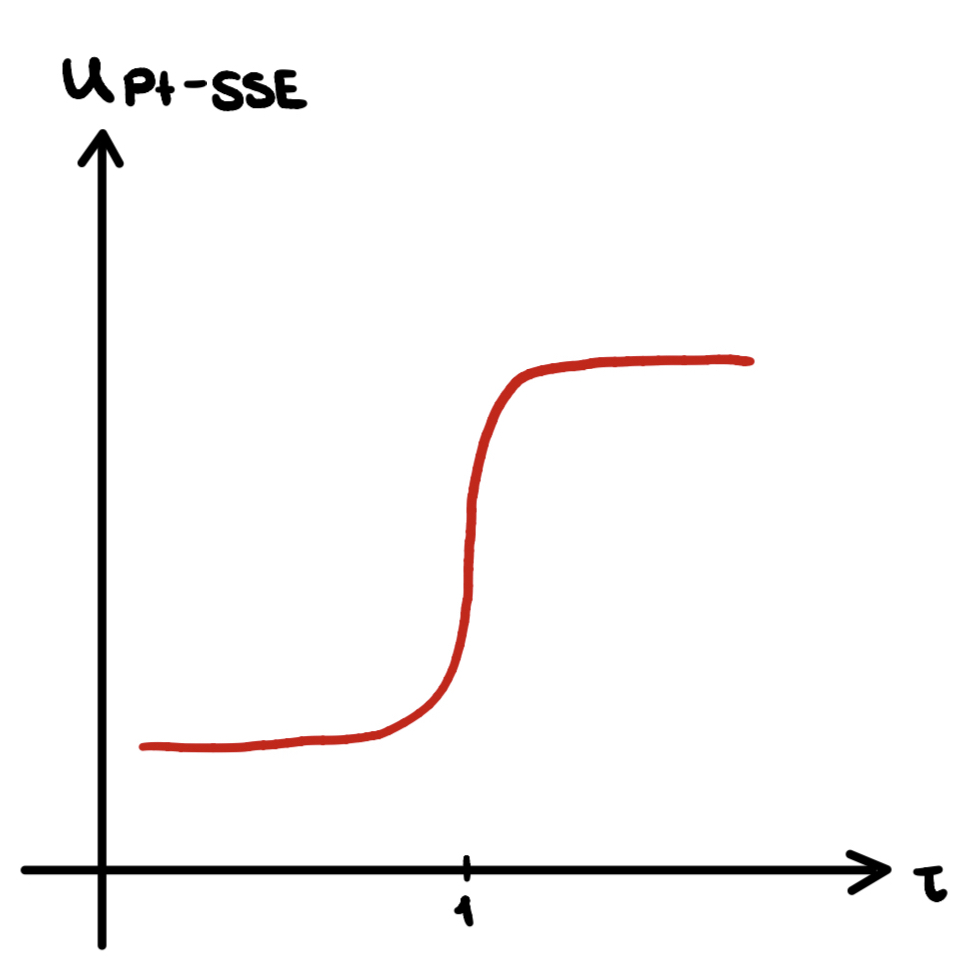
In der Potentiometrie wird die Potentialdifferenz im stromlosen Zustand gemessen. Dabei werden Bezugselektrode und Indikatorelektrode zu einer Messkette verbunden und die Potentialdifferenz zwischen beiden Elektroden gemessen. Da die Bezugselektrode ein konstantes Potential aufweist und die Indikatorelektrode, je nach Konzentration des Analyten, ein variables, ist die Potentialdifferenz proportional zur Konzentration des Analyten. Man kann einen Platin-Draht als Indikatorelektrode und eine Silber-Silberchlorid-Elektrode (SSE) als Bezugselektrode kombinieren. Es ist allerdings zu beachten, dass die Bezugselektrode keine gesättigte Kaliumchlorid(KCl)-Lösung enthält, sondern z.B. eine gesättigte Lithiumchlorid(LiCl)-Lösung in Ethanol. Dies liegt daran, dass durch die gesättigte KCl-Lösung auch Wasser in der Bezugselektrode wäre, was in die Probelösung gelangen könnte und so das Ergebnis beeinflussen würde. Da KCl aber nicht gut löslich ist in Ethanol, wird LiCl verwendet. Es ist trotzdem eine konstante Chlorid-Ionen-Konzentration innerhalb der Elektrode gegeben, die ausschlaggebend für das konstante Potential ist. Die durch diese Methode entstandene Kurve hat ein klassisches Aussehen, steigend von einem geringen Potential zu einem hohen (siehe Abbildung), denn mit Erreichen des Äquivalenzpunktes liegen freies Iod und Iodid vor, welche gemäß der Nernst-Gleichung das Potential in der Untersuchungslösung messbar verändern. Durch verschiedene Auswertungsverfahren kann der Äquivalenzpunkt innerhalb der Kurve bestimmt und so der Verbrauch abgelesen werden. 22
Phasendiagramm: ⚠ $Pt | Ox., Red. || Cl⁻, AgCl | Ag$
Coulometrisches Verfahren
In der Coulometrie wird der Gehalt des Analyten anhand der verbrauchten Ladungsmenge bestimmt. Dies ist möglich, da die für den Umsatz benötigte Ladung proportional zu der Masse des Analyten ist. Bei der coulometrischen Karl-Fischer-Titration wird durch elektrochemische Oxidation Iod erzeugt, welches dann als Edukt in die Maßreaktion eingeht.
Hier wird die galvanostatische Coulometrie verwendet. Bei dieser Variante wird die Elektrolysestromstärke konstant gehalten und der Strom tritt als Reagenz auf. Die benötigte Ladung kann daher aus der Stromstärke und der gemessenen Zeit bis zum Äquivalenzpunkt berechnet werden (⚠ $ { Q = I ⋅ t } $).
Da die Konzentration des Analyten im Laufe der Reaktion abnimmt und der Widerstand der Lösung damit zunimmt, erhöht sich die Spannung, gemäß ⚠ $ R = \frac{ U }{ I } $ . Diese erhöhte Spannung begünstigt Nebenreaktionen, was durch den Einsatz von Zwischenstoffen zum Abfangen dieser Spannung verhindert werden kann. Gleichzeitig wird dadurch ein Reagenz erzeugt, welches als Titrator des Analyten dienen kann.
Analog dazu funktioniert auch die Karl-Fischer-Titration. Es wird eine Iodid-haltige Lösung als Zwischenstoff verwendet. An der Anode entsteht dann durch Oxidation, bei relativ geringer Spannung, Iod.
Spannungsabfangreaktion und Reagenzerzeugung: 2I⁻ → I2 + 2e⁻
Dieses Iod kann dann als Maßreagenz in die Reaktion eingehen und Wasser aus dem Analyten umsetzen. Die Indikation erfolgt dabei entweder bivoltametrisch oder biamperometrisch. Bei beiden Indikationsverfahren wird ausgenutzt, dass nach dem Endpunkt ein reversibles Redoxsystem vorliegt. Das an der Anode entstandene Iod wird bei der Bestimmung von Wasser immer wieder zu Iodid reduziert. Sobald kein Wasser mehr vorhanden ist und das Iod, welches weiterhin an der Anode entsteht, nicht mehr reduziert werden kann, liegen Iod und Iodid gemeinsam in Lösung vor und bilden ein reversibles Redoxsystem. Dies zeigt sich bei der Biamperometrie durch einen Stromfluss und bei der Bivoltametrie durch einen Spannungsabfall am Äquivalenzpunkt. Vorteile dieser Variante sind die hohe Präzision des Verfahrens, es muss keine Maßlösung eingestellt oder gelagert werden, was Zeit und Platz spart und die Bestimmungsgrenze ist sehr gering, weswegen schon Bestimmungen im µg-Bereich möglich sind. 23
Instrumenteller Aufbau

Die Apparatur zur Titration besteht aus einem Titrationsgefäß (siehe Nr. 1 in Abb.) mit einer Doppelplatinelektrode (2), welche an eine Spannungsquelle angeschlossen ist. Es verfügt über zwei dicht verschließbare Öffnungen zur Zugabe von Lösungsmittel und Maßlösung (3). Eine weitere Öffnung dient zum Einlass von Luft (4) 25 und ist mit einem Molsieb (5) 26 zum Binden der Luftfeuchtigkeit versehen. Zusätzlich ist eine Öffnung zur Hinzugabe der Probe (6), welche entweder mit einem Stopfen oder einem Septum bei einer flüssigen Probe verschlossen ist, vorhanden. Gegebenenfalls gibt es auch weitere Ausstattungen wie eine Öffnung zur Zugabe von trockenem Stickstoff oder zum Absaugen von Lösungsmitteln. Der Titrierautomat führt die Endpunktbestimmung in der Regel automatisch durch. 27
Coulometrische Bestimmung:
Hierbei enthält der Titrierautomat eine Generatorelektrode, welche sich entweder direkt im Reaktionsraum befindet oder durch ein Diaphragma vom Reaktionsraum getrennt ist. Die Generatorelektrode erzeugt durch Oxidation von Iodid elektrochemisch Iod (siehe 1.3.2.4). 28
Durchführung
Die Karl-Fischer-Titration wird laut Arzneibuch nach den Vorgaben des Geräte-Herstellers durchgeführt. 29
Die Titration kann volumetrisch oder coulometrisch erfolgen (siehe 1.3.2). 30
Zur Titration wird das Karl-Fischer-Reagenz verwendet, welches das zur Bestimmung notwenidige Iod und Schwefeldioxid sowie die Hilfsbase, gelöst in einem wasserfreien Medium, enthält. Das verwendete wasserfreie Medium ist entweder Methanol R, das in der Monographie angegebene Lösungsmittel, oder das vom Gerätehersteller vorgeschlagene Lösungsmittel. 31
Bestimmung des Wirkwerts
Vor der Durchführung der eigentlichen Titration muss zunächst der Wirkwert bestimmt werden, da die umgesetzte Menge Wasser mit Iod nicht der eigentlichen Menge Wasser in der Probe entspricht. In den Lösemitteln und Reagenzien kann auch Wasser enthalten sein, welches dann schon vor Beginn der Titration Iod umsetzt. Auch Luftfeuchtigkeit trägt zu diesem Prozess bei. 32
Für die Bestimmung wird Methanol R oder das vom Hersteller der Maßlösung vorgeschlagene Lösungsmittel verwendet. Das Restwasser muss vor der Titration aus dem Probengefäß / der Messzelle entfernt werden oder es findet eine Vortitration statt. 33 Bei einer Vortitration wird das Lösungsmittel mit dem Karl-Fischer-Reagenz bis zum Äquivalenzpunkt titriert, dementsprechend wird das vorhandene Wasser nach der Karl-Fischer-Reaktion (siehe 1.3.1) umgesetzt. Dieser Vorgang wird auch als Konditionierung bezeichnet. Dem konditionierten Ansatz wird nun eine bekannte Menge Wasser R oder der vorgegebene Standard mit bekanntem Wassergehalt hinzugefügt. Danach kann die Titration zur Bestimmung des Wirkwerts unter kontinuierlichem Rühren der Lösung durchgeführt werden. 34
Berechnet wird der Wirkwert nach folgender Formel 35:
⚠ $$Wirkwert = \frac{x\ mg \quad H_{2}O}{1ml \quad KF-Reagenz}⚠ $$
Der bestimmte Wirkwert wird als Wasseräquivalent angegeben, also wie viel mg Wasser durch 1 mL Karl-Fischer-Reagenz umgesetzt wurden. 36 37 Dieser muss nach Arzneibuchvorgaben mindestens 80% des auf der Maßlösung angegebenen Wertes betragen. Diese Bestimmung wird nicht nur vor der ersten Titration sondern auch in regelmäßigen Abständen zwischen den folgenden Titrationen wiederholt. 38
Vermessen der Probe
Anschließend kann die Titration der Probe erfolgen. Auch hierbei muss Restwasser vor der Titration aus dem Probengefäß / der Messzelle entfernt werden oder eine Vortitration durchgeführt werden. 39 Die Substanz wird nach Vorbereitung des Lösemittels zügig hinzugegeben. 40 Da es sich bei der Karl-Fischer-Titration um eine Bestimmung des Wassergehalts handelt, muss während der Titration und bei der Probenvorbereitung strengstens darauf geachtet werden, dass unter Wasserausschluss gearbeitet wird. 41 Nebenreaktionen, welche Wasser erzeugen, müssen vermieden werden. 42 Auch die Probe und die verwendeten Reagenzien dürfen vor der Durchführung der Titration nicht der Luftfeuchtigkeit ausgesetzt sein. 43
Die Zutitration erfolgt mittels Mikroprozessor-gesteuerter Kolbenpumpen. 44 Der Endpunkt kann unterschiedlich detektiert werden. Zum einen kann ein Farbumschlag am Endpunkt visuell oder photometrisch erkannt werden, zum anderen können elektrochemische Indikationen eingesetzt werden (siehe 1.3.2). 45
Je nach erwartetem Reaktionsverlauf wird nach der Methode A oder der Methode B gearbeitet.
Methode A
Es wird eine direkte Titration durchgeführt. Diese Methode wird als Standard angewendet. 46 Während der Reaktion wird die Probelösung kontinuierlich gerührt, dies geschieht mit Hilfe eines Magnetrührers. 47 Angewendet wird die Methode nur, wenn die Reaktion genügend schnell abläuft und der Endpunkt genau genug zu erkennen ist. 48
Methode B
Als Alternative zu Methode A wird Methode B eingesetzt, wenn die Reaktionsgeschwindigkeit nicht hoch genug ist oder der Endpunkt nicht klar zu erkennen ist. Die Probe wird mit Maßlösung im Überschuss versetzt, um eine Rücktitration durchzuführen. Die überschüssige Maßlösung kann dann mit Methanol R oder dem vorgeschriebenen Lösungsmittel rücktitriert werden. 49 50
Einzelnachweise
1 Fischer, Karl: Neues Verfahren zur maßanalytischen Bestimmung des Wassergehaltes von Flüssigkeiten und festen Körpern. (31.05.1935), URL: https://onlinelibrary.wiley.com/doi/pdf/10.1002/ange.19350482605 (Stand: 05.06.2021) ⇑
2 Janaik, Christoph/ Zuelsdorf, Sabine: Karl-Fischer Titration (26.03.2010), URL: https://www.instmeth.uni-freiburg.de/methods/iaac/rk/m-kf-titration.pdf (Stand: 05.06.2021) ⇑
3 Unbekant: Wasserbestimmung mit Karl-Fischer-Titration (XX.XX.XXXX), URL: https://www.ipc.uni-jena.de/ipcmedia/lehre/iaii/iaii_12_karl-fischer-titration.pdf (Stand: 05.06.2021) ⇑
4 E. Ehlers: Analytik II; 11. Auflage; Deutscher Apotheker Verlag, Stuttgart 2008, S.209f ⇑
5 Eigene Abbildung nach Scholz, Eugen: Karl-Fischer-Titration (XX.XX.1984), URL: https://link.springer.com/chapter/10.1007/978-3-642-69368-7_2(Stand: 05.06.2021) ⇑
6 E. Ehlers: Analytik II; 11. Auflage; Deutscher Apotheker Verlag, Stuttgart 2008, S.209f ⇑
7 G. Rücker/M. Neugebauer/G. G. Willems: Instrum. pharm. Analytik; 5. Auflage; Wiss. Verlagsgesell., Stuttgart 2013, S.662 ⇑
8 Unbekannt: Karl-Fischer-Verfahren (XX.XX.XXXX), URL: https://www.chemie.de/lexikon/Karl-Fischer-Verfahren.html (Stand: 05.06.2021) ⇑
9 E. Ehlers: Analytik II; 11. Auflage; Deutscher Apotheker Verlag, Stuttgart 2008, S.209f ⇑
10 Eigene Abbildung nach Unbekannt: Karl-Fischer-Verfahren (XX.XX.XXXX), URL: https://www.chemie.de/lexikon/Karl-Fischer-Verfahren.html (Stand: 05.06.2021) ⇑
11 Unbekannt: Karl-Fischer-Titration (XX.XX.XXXX), URL: http://www.titrationen.de/karl-fischer-titration/ (Stand: 05.06.2021) ⇑
12 Unbekannt: Karl-Fischer-Titration - Wie Erkennt man Nebenreaktionen? (XX.XX.XXXX), URL: https://metrohm.com/~/media/metrohm%20germany/downloads/flyer/f_kf_0205_v1_16-wie-erkennt-man-nebenreaktionen-bei-der-kft-web.pdf?la (Stand: 05.06.2021) ⇑
13 Eigene Abbildung nach: Einführung in die Instrumentelle Analytik: Elektrochemische Methoden, Orban, O., 2020, S. 100 ⇑
14 Dickenhorst, Burkhard: Biamperometrie (2001), URL: http://www.bdsoft.de/demo/index.htm?/demo/chemie/analytik/elektrochemisch/biamperometrie.htm (Stand: 30.05.2021) ⇑
15 Unbekannt: Voltammetrie (XXXX), URL: https://de.wikipedia.org/wiki/Voltammetrie (Stand: 19.06.2021) ⇑
16 Dickenhorst, Burkhard: Biamperometrie (2001), URL: http://www.bdsoft.de/demo/index.htm?/demo/chemie/analytik/elektrochemisch/biamperometrie.htm (Stand: 30.05.2021) ⇑
17 Einführung in die Instrumentelle Analytik: Elektrochemische Methoden, Orban, O., 2020, S. 100 ⇑
18 Eigene Abbildung nach: Einführung in die Instrumentelle Analytik: Elektrochemische Methoden, Orban, O., 2020, S. 100 ⇑
19 Dickenhorst, Burkhard: Bivoltametrie (2001), URL: http://www.bdsoft.de/demo/index.htm?/demo/chemie/analytik/elektrochemisch/biamperometrie.htm (Stand: 30.05.2021) ⇑
20 Einführung in die Instrumentelle Analytik: Elektrochemische Methoden, Orban, O., 2020, S. 100 ⇑
21 Eigene Abbildung nach: Einführung in die Instrumentelle Analytik: Elektrochemische Methoden, Orban, O., 2020, S. 52 ⇑
22 Einführung in die Instrumentelle Analytik: Elektrochemische Methoden, Orban, O., 2020, S. 24, 34, 37, 40, 99 ⇑
23 Einführung in die Instrumentelle Analytik: Elektrochemische Methoden, Orban, O., 2020, S. 102, 105, 106, 107 ⇑
24 Eigene Aufnahme, Beschriftung nach: Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
25 Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
26 Einführung in die Instrumentelle Analytik: Elektrochemische Methoden, Orban, O., 2020, S. 100 ⇑
27 Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
28 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
29 Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
30 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
31 Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
32 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
33 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
34 Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
35 Einführung in die Instrumentelle Analytik: Elektrochemische Methoden, Dr. O. Orban, 2020, S.99 ⇑
36 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
37 Einführung in die Instrumentelle Analytik: Elektrochemische Methoden, Dr. O. Orban, 2020, S.99 ⇑
38 Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
39 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
40 Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
41 Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
42 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
43 Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
44 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
45 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
46 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
47 Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
48 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
49 Kommentar zur Ph. Eur. 9.4/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
50 Ph. Eur. 10.0/2.05.12.00 Halbmikrobestimmung von Wasser – Karl-Fischer-Methode ⇑
Monographiebeispiel: Omeprazol-Natrium, Prüfung auf Reinheit, Gehalt an Wasser
Stoffcharakterisierung
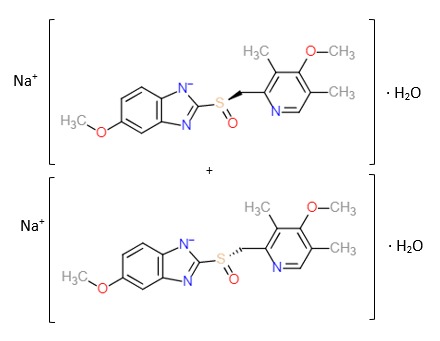
Omeprazol-Natrium trägt den IUPAC-Namen Natrium[5-methoxy-2-[(RS)-[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl]-1H-benzimidazol]-Monohydrat, hat eine relative Molare Masse von 385,4 und besitzt die Summenformel C17H18N3NaO3S ⋅ H2O, weshalb es demnach als Monohydrat vorliegt. Bei diesem Stoff handelt es sich um ein Enantiomerengemisch.
Das Arzneibuch stellt eine Gehaltsanforderung von 98,0 % bis 101,0 %, wobei hier die wasserfreie Substanz betrachtet wird.
Omeprazol-Natrium ist ein weißes bis fast weißes, hygroskopisches Pulver und zudem leicht löslich in Wasser und Ethanol 96%. In Propylenglycol ist es löslich und in Dichlormethan nur sehr schwer löslich. 2
Der Wirkstoff zählt zur Gruppe der Protonenpumpeninhibitoren, kurz PPI. Als Prodrug in magensaftresistenter Form, über oralem Wege aufgenommen, wird das Omeprazol im Darm resorbiert und mit dem Blut zu den Belegzellen der Magenschleimhaut transportiert. Dort wird es durch den sauren pH-Wert in seine Wirkform, die Sulfenamide, umgewandelt. 3 Durch die Sulfenamide kommt es zu einer irreversiblen Hemmung der Protonen-Kalium-ATPasen der Magensäure-produzierenden Belegzellen, was zu einer Verringerung der Magensäureproduktion führt. 4 So kommt es also zum Anstieg des pH-Werts im Magen und die schützende Schleimschicht kann durch die Magenschleimhautzellen wieder aufgebaut werden.
Aus diesem Grund wird Omeprazol bei Erkrankungen des Gastrointestinaltraktes, die durch die Magensäure gefördert werden, wie zum Beispiel bei Magengeschwüren / Darmgeschwüren (Ulcus ventriculi / duodeni) oder gastroösophagealen Refluxkrankheiten, eingesetzt. Ebenso wird es bei Infektionen mit dem Magenbakterium Helicobacter pylori, welches zur Steigerung der Säureproduktion führt oder als Prophylaxe bei Einnahme der Magensäureproduktion-steigernden nichtsteroidalen Antirheumatika wie z.B. Acetylsalicylsäure oder Ibuprofen, angewendet. 5
Durchführung der Monographie
Das Arzneibuch lässt unter der Prüfung auf Reinheit einen Wassergehalt von 4,5 % bis 10,0 %, bei Bestimmung von 0,300 g Substanz zu. Für diese Prüfung wird die Halbmikrobestimmung von Wasser nach der Karl-Fischer-Methode (2.5.12) vorgeschrieben. Das genaue Vorgehen ist bereits in Absatz 1.5 beschrieben worden.
Eignung der analytischen Methode
Da das Lösungsmittel für die Karl-Fischer-Titration von Hersteller zu Hersteller unterschiedlich sein kann, weil das Arzneibuch kein spezifisches Lösungsmittel vorschreibt, ist für jede Kombination von Analysensubstanz, Maßlösung und Lösungsmittel eine spezifische Prüfung erforderlich. Damit soll sichergestellt werden, dass die verwendeten Lösungen zur Bestimmung des Wassergehalts der Substanz im erforderlichen Maße geeignet sind. Die sogenannte Eignungsprüfung ist laut Arzneibuch bei Substanzen mit einem Wassergehalt von 2,5 mg bis 25 mg einsetzbar. Bei Vermessung von 0,300 g Omeprazol-Natrium und einem zugelassenen Wassergehalt von 4,5 % bis 10,0 %, sind es hier 13,5 mg bis 30 mg Wasser. Demnach ist die Eignungsprüfung hier in einem großen Bereich anwendbar.
Für die Eignungsprüfung wird die Analysensubstanz mit dem gewählten Lösungsmittel titriert und so der Wassergehalt bestimmt. Danach werden in mindestens fünf Zusätzen jeweils bekannte Wassermengen, die etwa 50% bis 100 % der in der Analysensubstanz enthaltenen Wassermenge entsprechen, zugesetzt und nach jedem Zusatz der Wassergehalt bestimmt. Für jeden Zusatz wird die Wiederfindung (r, in %) bestimmt, welche wie folgt berechnet wird:
⚠ $$ r = \frac{ 100 ⋅ W_{ 2 } }{ W_{ 1 } } ⚠ $$
Dabei steht W1 für die Menge des zugesetzten Wassers (in mg) und W2 für die bestimmte Menge des Wassers (in mg). Aus allen Wiederfindungen wird die mittlere Wiederfindung ( ⚠ $ \overline{r} $ ) berechnet. Solange ⚠ $ \overline{r} $ zwischen 97,5 % bis 102,5 % liegt, gilt die Kombination von Substanz, Maßlösung und Lösungsmittel als geeignet. Aus den nun gegebenen Werten wird eine Regressionsgerade erstellt, wobei auf der x-Achse die zugefügten Wassermengen und auf der y-Achse die bestimmten Wassermengen (M in mg) aufgetragen werden. Anschließend wird der Schnittpunkt mit der y-Achse (a in mg), die Steigung (b) und der Schnittpunkt der Kalibriergeraden mit der x-Achse (d in mg) errechnet. Daraufhin wird die Fehlerquote (e1 und e2, in %) bestimmt. Dies geschieht durch folgende Gleichungen:
⚠ $$ e_{ 1 } = \frac{ 100 ⋅ (a - M) }{ M } ⚠ $$
⚠ $$ e_{ 2 } = \frac{ 100 ⋅ (|d| - M) }{ M } ⚠ $$
Solange auch e1 und e2 maximal 2,5 % betragen und b zwischen 0,975 und 1,025 liegt, ist die Substanz-Maßlösung-Lösungsmittelkombination geeignet zur Wasserbestimmung durch die Karl-Fischer-Methode. 6
Einzelnachweise
1 Eigene Abbildung nach Ph. Eur. 10.0/1032: Monographie Omeprazol-Natrium ⇑
2 Ph. Eur. 10.0/1032: Monographie Omeprazol-Natrium ⇑
3 Klein, Sonja: Omeprazol (06.02.19), https://www.gelbe-liste.de/wirkstoffe/Omeprazol_453 (Stand: 05.06.21) ⇑
4 Dr. Moser, Gerd: Omeprazol, https://www.pharmazeutische-zeitung.de/omeprazol/ (Stand: 05.06.21) ⇑
5 Klein, Sonja: Omeprazol (06.02.19), https://www.gelbe-liste.de/wirkstoffe/Omeprazol_453 (Stand: 05.06.21) ⇑
6 Ph. Eur 10.0/ Monographie 2.05.12.00: Halbmikrobestimmung von Wasser - Karl-Fischer-Methode ⇑
TU-Braunschweig Institut für Medizinische und Pharmazeutische Chemie Seminar: Instrumentelle Analytik Kontakt: tubs@t-kellner.de
