Gaschromatographie
Bericht der Expertengruppe für
Gaschromatographie
SoSe 2021
Abgabedatum
21.06.2021
Über-/Expertengruppe 04
Autorinnen:
Tatiana Gorpishina
Marie Rasemann
Charlotte Kalinwoski
Esma Kurt
Melina Hüske
Wiebke Heitmann
Gaschromatographie
Inhaltsverzeichnis
Einleitung
Die Gaschromatographie (GC) ist ein physikalisches Trennverfahren, welche einen Analyten mithilfe von Wechselwirkungen mit der stationären Phase trennen kann. Abhängig von der stationären Phase erfolgt die Trennung entweder nach dem Verteilungsprinzip (flüssige stationäre Phase) oder dem Absorptionsprinzip (feste stationäre Phase).1 Sie eignet sich zur qualitativen und quantitativen Analyse von Stoffgemischen.2 Dabei lassen sich die Stoffgemische vom Trägergas, welches die mobile Gasphase ist, überführen. Zu beachten ist hierbei, dass keine Interaktion weder mit dem Analyt, noch mit der stationären Phase stattfinden darf. Aus diesem Grund werden Gase verwendet, die nicht mit der stationären Phase wechselwirken (Inertgase). Das Ergebnis einer gaschromatographischen Analyse wird als Gaschromatogramm bezeichnet.
Physikalische und chemische Grundlagen
Der Gaschromatograph ist aufgebaut aus:
- Injektor, der die Probe aufgibt
- Ofen mit Trennsäule
- Detektor
Als erstes wird die zu analysierende Substanz in den Injektor gegeben, wo sie bei bis zu 450 °C in den Trägergasstrom eingebracht wird und in die Gasphase übergeht. Wichtig dabei ist, dass die Substanzen im Injektor verdampft werden, wenn sie nicht gasförmig vorliegen. Daher eignen sich Stoffe, die bereits gasförmig sind oder unzersetzt in die Gasphase überführt werden können.3 Durch eine Derivatisierungsreaktion (Reaktion polarer Gruppen mit verschiedenen Reagenzien, sodass der Siedepunkt erniedrigt wird) können schwer flüchtige Substanzen, wie zum Beispiel Zucker, in flüchtige Verbindungen überführt werden.4 Als nächstes werden die verdampften bzw. gasförmigen Substanzen mit dem Trägergasstrom durch die Trennsäule transportiert, wobei der Ofen auf bis zu 350 °C erhitzt werden kann. Durch eine Verteilung zwischen der mobilen (Trägergasstrom) und der stationären Phase (Trennflüssigkeit) erfolgt die Substanztrennung. Anhand eines Detektors werden die eluierenden Substanzen, also die absorbierten Stoffe, die aus dem Adsorptionsmittel herausgelöst werden, detektiert.5
Je nachdem welcher Detektor verwendet wird, wird ein passendes Trägergas gewählt: Häufig orientiert sich die Auswahl an kurzen Analysezeiten sowie an höhere Strömungsgeschwindigkeiten, weshalb beispielsweise bei gepackten Säulen Stickstoff als Trägergas in Frage kommt und bei den Kapillarsäulen Helium oder Wasserstoff.6
Außerdem spielt die Temperatur eine wichtige Rolle. Je niedriger die Temperatur ist, umso länger verbleibt die Substanz auf der Trennsäule und umso weiter sind die Peakmaxima voneinander entfernt. Das liegt daran, dass Gase bei niedrigeren Temperaturen besser in Flüssigkeiten löslich sind. Bei komplexen Gemischen kann ein Temperaturprogramm verwendet werden (konstantes Aufheizen), da sich große Flüchtigkeitsbereiche ansonsten nicht optimal trennen lassen. Als Retentionszeit (tR) wird die Zeit bezeichnet, die eine Substanz vom Injektor bis zum Detektor benötigt. Je mehr die Substanz mit der stationären Phase wechselwirkt, umso länger ist die Retentionszeit. Die unterschiedlichen Trennzeiten der Komponenten ergeben somit ein Gaschromatogramm.8
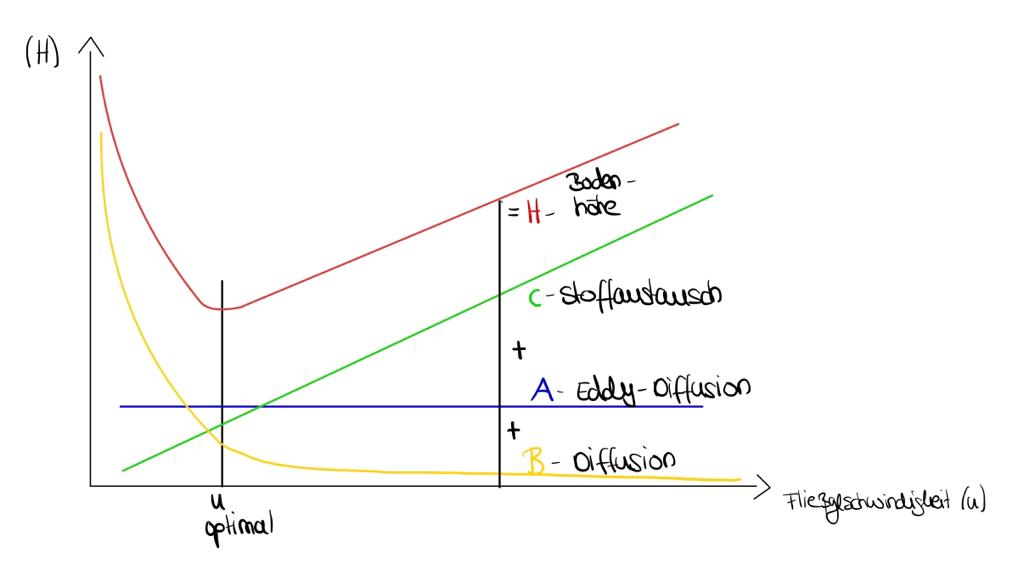
Für die Ermittlung der optimalen Fließgeschwindigkeit zieht man die Van-Deemter-Kurve heran. Sie beschreibt den Einfluss der Trägergasgeschwindigkeit (u) auf die Trennleistung einer Säule. Dafür trägt man die Trennstufenhöhe gegen die Fließgeschwindigkeit (u) auf (siehe Abbildung).
Mit der abgebildeten Gleichung (siehe rechts) lässt sich die Bodenhöhe (H) ermitteln.
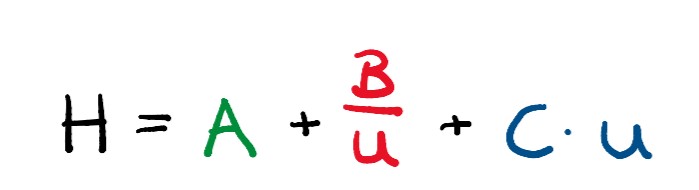
Der Term A gibt die Eddy-Diffusion an, welche unterschiedliche Flusswege einzelner Probemoleküle durch die Säule beschreibt. Dieser ist unabhängig von der Fließgeschwindigkeit (u). Die Longitudinal-Diffusion wird von dem Term B angegeben und ist antiproportional zu u. Denn je größer die Fließgeschwindigkeit ist, umso geringer ist die Tendenz des Moleküls sich seitlich zu bewegen. Im Gegensatz dazu ist Term C, welcher den Massenübergang zwischen stationärer und mobiler Phase beschreibt, proportional zur Fließgeschwindigkeit. 10 Die optimale Fließgeschwindigkeit (u) hat ein Minimum, wenn die Bodenhöhe (H) am geringsten ist, wodurch die Trennleistung am größten ist.11, 12
Ein wichtiger Aspekt der Gaschromatographie ist der Kovats-Index, welchen man zur Auswertung heranzieht. Dieser ermöglicht das Ausgleichen der Retentionszeiten. Dabei wird die Beziehung zwischen der Retentionszeit und dem Kohlenstoffgehalt genutzt, um anschließend einen geräteunabhängigen Wert für die Identifikation von Substanzen zu erhalten. Definiert ist der Index der n-Alkanen als ihre Kettenlänge x100. Hier wird der Logarithmus der Nettoretentionszeiten gegen den Index aufgetragen, wobei der unbekannte Index interpoliert wird, da dieser abhängig von der stationären Phase und der Temperatur ist. 13
Quantitative Bestimmung
Voraussetzung für eine quantitative Bestimmung ist, dass der Detektor in einem linearen Konzentrationsbereich arbeitet, wofür der interne Standard herangezogen wird. Dafür werden Lösungen mit verschiedenen Konzentrationen auf die durch den internen Standard korrigierte Peakfläche injiziert. Die Peakfläche, welche ein wichtiger Faktor für die quantitative Bestimmung ist, ist proportional zu der Konzentration bzw. der Masse der Probelösung.14 Der interne Standard ist grundlegend wichtig, da diese als eine relative Bezugsgröße dient, welcher den Einfluss des Verfahrens auf das Ergebnis abbilden soll. Auf diese Weise wird eine Einschätzung der Qualität ermöglicht. Es handelt sich hierbei um Substanzen, die dem Analyten möglichst ähnlich sind, in der Analysenprobe jedoch nicht vorkommen. Der interne Standard wird in gleichen molaren Verhältnis zu den Analysenlösungen zugegeben wird. Das Ziel ist es, Dosierfehler zu korrigieren. 15 Die Kalibrierung ist die Bestimmung der Abhängigkeit zwischen Substanzkonzentrationen und dem Messsignal. Sie wird durchgeführt, um die Konzentration der Analyten zu bestimmen. Der Responsefaktor ist die Steigung der Kalibriergeraden und dient zur Ermittlung der Empfindlichkeit des Verfahrens.
Qualitative Bestimmung
Das Grundprinzip zur Auswahl des Säulenmaterials besteht darin, dass unpolare Stoffe an unpolaren Trennphasen getrennt werden. Die am häufigsten verwendete Trennphase ist Methylpolysiloxan. Allgemein gibt es eine Regel zur Bestimmung der Elutionsreihenfolge, die besagt, dass der Siedepunkt einer Substanz mit den Van-der-Waals-Kräften korrelieren kann. Je länger eine Seitenkette ist, umso stärker sind die Van-der-Waals-Kräfte. Dementsprechend sind die Wechselwirkungen zwischen dem Molekül und der Trennschicht höher. Daraus folgt, dass die Retentionszeit mit wachsender Kettenlänge zunimmt. Bei einer homologen Reihe mit verstärkter Verzweigung nimmt die Retention ab, da hierbei die Van-der-Waals-Kräfte sowie die Siedepunkte sinken, wohingegen der Dampfdruck steigt. Somit lassen sich Substanzen anhand der Retentionszeiten zuordnen. 16
Vorteile und Nachteile
Vorteile:
- die Komponenten können bei der GC sowohl identifiziert als auch quantifiziert werden17
- drastisch bessere Auftrennung ähnlicher Stoffe möglich18
- das Gleichgewicht zwischen Trägergas und stationärer Phase stellt sich schnell ein, was hohe Trägergasgeschwindigkeiten und somit eine hohe Trenneffizienz ermöglicht 19
- der Zeitaufwand für die Analysen wird vermindert, da immer dünnere und kürzere Säulen verwendet werden 20
- geringer Substanzbedarf erforderlich (im Mikrogramm Bereich und im oberen Nanogramm Bereich) 21
Nachteile:
- weniger geeignet für die Isolierung von Substanzen 22
- viele der Zielsubstanzen sind ohne vorherige Derivatisierung kaum bzw. schlecht gaschromatographierbar, da sie häufig z.B. thermisch labil sind und sich zersetzen können 23
Instrumenteller Aufbau
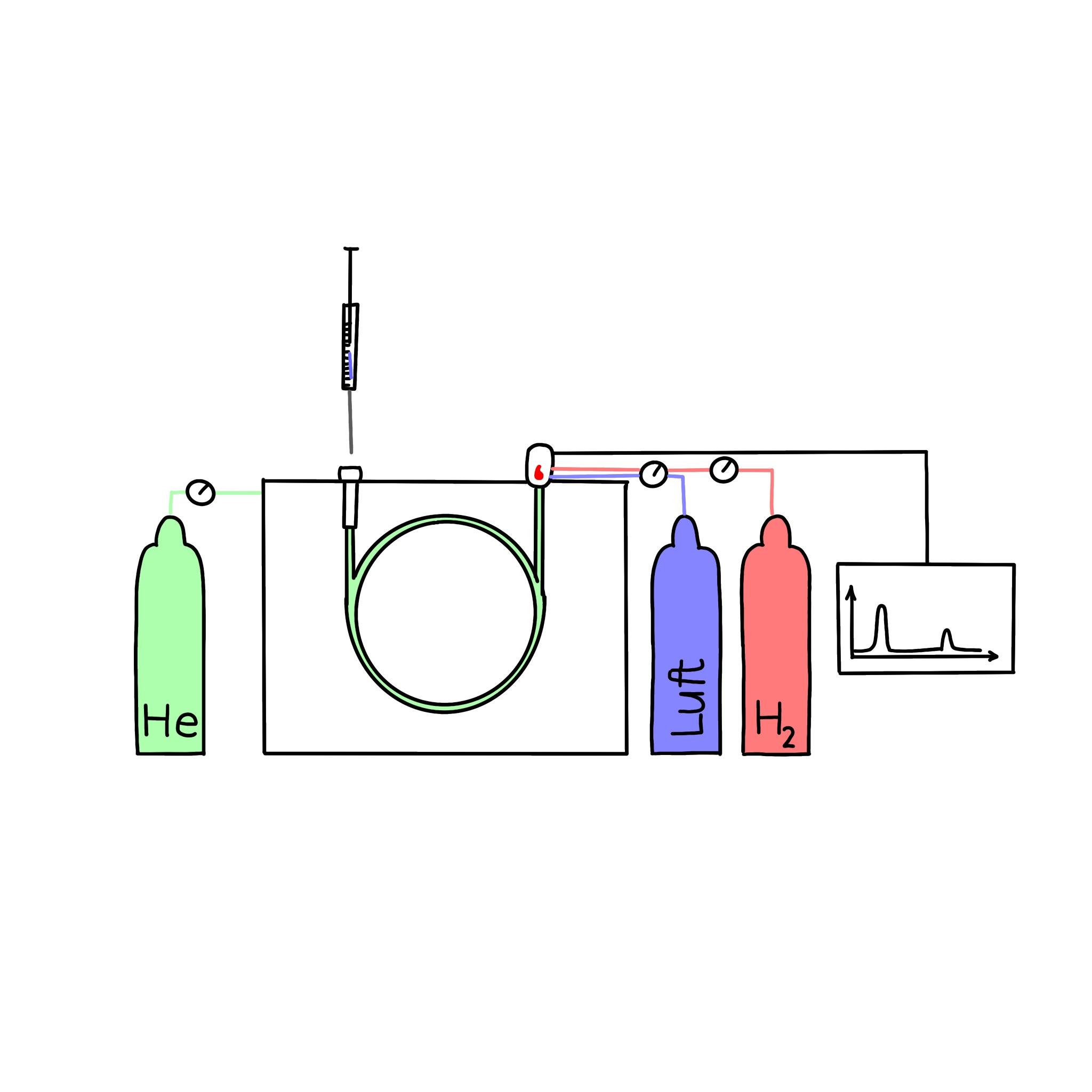
Ein Gaschromatograph besteht im Allgemeinen aus einer Gasdruckflasche, einem Injektor, einem Säulenofen mit einer darin befindlichen Säule, einem Detektor und einem Schreiber.25 Die einzelnen Bestandteile werden im nachfolgenden Kapitel genauer erläutert.
Gasdruckflasche
Die Gasdruckflasche liefert das Gas, welches die mobile Phase darstellt. Es wird auch als Trägergas bezeichnet. Helium, Wasserstoff, Argon und Stickstoff kommen hierbei in Frage und müssen besondere Anforderungen erfüllen.26
Ein Kriterium ist, dass die Trägergase hochrein sein müssen, das heißt es wird gewährleistet, dass keine weiteren Substanzen (Verunreinigungen) im Trägergas enthalten sind, die mit der stationären Phase und dem Analyten wechselwirken könnten. Somit ist das Trägergas inert. Des Weiteren sollten sie sauerstoff- und wasserfrei sein. Wenn dies nicht der Fall ist, kann es zur Oxidation bzw. Hydratisierung der Teilchen der stationären Phase kommen, welche die Trenneffizienz verringern würde.27
An der Gasdruckflasche befinden sich ein Druckminderer und ein Feinregler, der eine konstante Durchströmung der stationären Phase mit dem Trägergas gewährleistet.28
Injektor
Der Injektor überführt den Analyten, wenn dieser nicht bereits gasförmig ist, vom flüssigen in den gasförmigen Aggregatzustand, welcher dann auf die stationäre Phase transportiert wird.
Die Wahl des Injektors hängt von der im Versuch verwendeten Säule ab. Demnach unterscheidet man in:
- Split-Injektor
- On-Column-Injektor
- Probenschleifen
- Temperaturprogrammierbarer Injektor
- Head-Space Analyse
- Direktinjektion (relativ obsolet)
Im Folgenden werden die wichtigsten Injektionsverfahren erläutert.
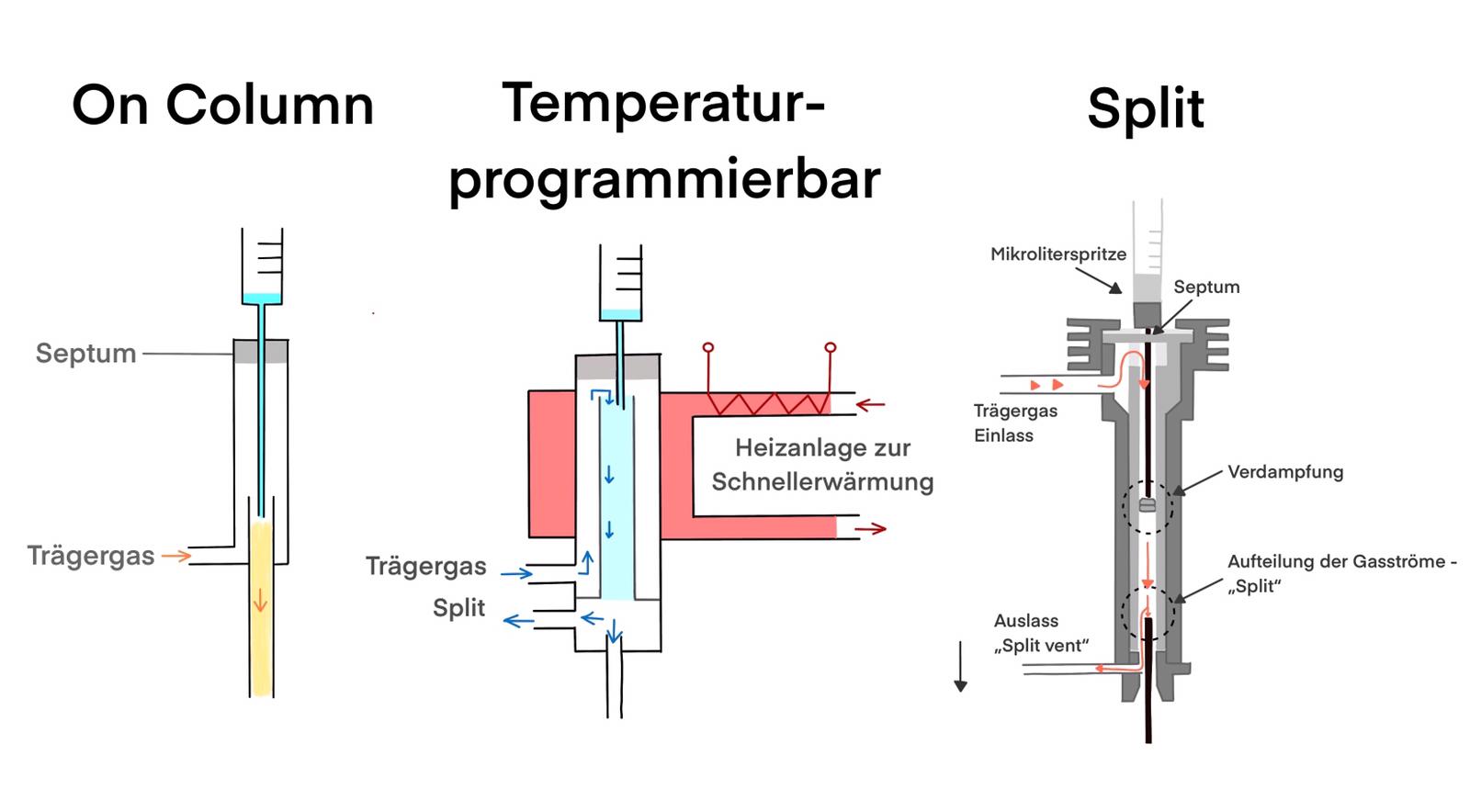
Die Splitinjektion
Beim Split-Injektor ist ein sogenannter Split vorhanden. Dieser kann offen oder geschlossen sein. Die flüssige Probe wird mithilfe einer Spritze durch ein luftdichtes Septum in den beheizten Probenraum gegeben. Die Temperatur des Probenraums wird je nach Probe, Trägergas und Säulenmaterial individuell gewählt. Dabei wird sie schlagartig in die Gasphase überführt. Durch den Split wird nur ein Teil der verdampften Probe auf die Säule gebracht, das heißt sie wird gesplittet. Die abgetrennte Menge an Probe, die nicht auf die Säule transportiert wird, gelangt in ein Abfallgefäß.30, 31 Dies verhindert das Überladen der Säule, welches sich durch abgeschnittene Peakspitzen und unsymmetrische Peaks äußern würde und die nachstehende quantitative Bestimmung der Probe erschwert. 32
Des Weiteren ist zu erwähnen, dass durch die Splitinjektion diskriminierende Effekte, bezogen auf die Probe, vorkommen können. Dies bedeutet, dass höher siedende Komponenten im Split verbleiben und niedrig siedende Substanzen bevorzugt in die Säule übergehen. Diskriminierende Effekte können mithilfe der Direktinjektion verhindert werden.33
On-Column-Injektor
Bei dem On-Column-Injektor-Verfahren handelt es sich um eine kalte Verfahrenstechnik, das heißt der Injektorraum wird nicht beheizt und gegebenenfalls gekühlt. Dabei wird die Probe nach der Injektion nicht sofort verdampft, sondern in flüssiger Form, mithilfe eines speziellen Injektionsverfahrens und einer Spritze mit dünner Nadel, in den Kapillarsäuleneingang injiziert. Der Säulenofen wird anschließend hochgeheizt und die Probe verdampft. Dieses Verfahren ist damit für thermolabile Stoffe geeignet und ermöglicht eine Aufkonzentration der Probe, welche durch das frühzeitige Verdampfen des Lösemittels erfolgt.34
Probenschleifen
Bei der Probenschleife wird ein eingestelltes, reproduzierbares Volumen auf die Säule injiziert. Somit können Versuche mit vergleichbaren Bedingungen wiederholt und automatisiert werden.35
Temperaturprogrammierbarer Injektor
Diese Methode eignet sich für Analysen, bei denen nur ausgewählte Substanzen in einer Probe analysiert werden sollen. Die Probe wird in einen kalten Probenraum injiziert und langsam durch eine Heizspirale aufgeheizt. Beim Erreichen einer bestimmten Temperatur (Siedetemperatur der zu analysierenden Substanz) wird der bis zu diesem Zeitpunkt geöffnete Split geschlossen. Dadurch befinden sich nur die Substanzen auf der Säule, welche in diesem Bereich sieden und somit exklusiv analysiert werden können.36 Aufgrund der schnellen Änderung der Temperatur kombiniert mit einem Split vereint der temperaturprogrammierbare Injektor die Vorteile des Split-Injektors und des On-Column-Injektors.
Head-Space Analyse
Diese Analysenmethode ist ein „Add-on“ zu anderen Injektionssystemen. Sie eignet sich für Analyten, bei denen leicht flüchtige Stoffe in einer Matrix vorliegen, z.B. bei Blutproben. Dabei soll die Matrix nicht ins chromatographische System gelangen.
Die Probe wird in ein „Head-Space-Vial“ gegeben. Dieses ist luftdicht verschlossen und kann gegebenenfalls noch erhitzt werden. Zwischen der flüssigen und gasförmigen Phase stellt sich ein Gleichgewicht ein, wobei sich die gasförmige Phase im oberen Bereich des Vials befindet. Mittels einer gasdichten Spritze wird eine bestimmte Menge der Gasphase, in der sich der zu analysierende Probenbestandteil befindet, entnommen und über einen Split-Injektor injiziert.37
Trennsäulen
Die Trennsäulen, die in der Gaschromatographie ihren Einsatz finden, lassen sich in gepackte Säulen und Kapillarsäulen unterteilen.38 Die Vorteile der gepackten Säulen sind ihre Robustheit und Kapazität, wohingegen Kapillarsäulen durch ihre hohe Trennleistung von großer Bedeutung sind.39
Die Trennflüssigkeiten können Squalan, alkylierte und acylierte Cyclodextrine, Polyethylenglykole und Methylpolysiloxane sein.40
Gepackte Säulen
Gepackte Säulen bestehen in der Regel aus einem Glas- bzw. Metallrohr. Sie können in Innendurchmesser (2 bis 4 Millimeter) und der Länge (1 bis 5 Meter, die auf eine Spule aufgewickelt sind) variieren.41 Die Säulen sind mit festen Adsorbentien oder Flüssigkeiten, die auf ein Trägermaterial aufgebracht sind, gefüllt. Demnach unterscheidet man in Gas-Fest-Chromatographie und Gas-Flüssig-Chromatographie.
Die Gas-Fest-Chromatographie wird auch Adsorptionschromatographie genannt. Als Adsorbentien kommen Kieselgel, Aluminium(III)-oxid, Aktivkohle und verschiedene organische Polymere in Frage. Sie findet häufig Anwendung bei der Bestimmung von Gasen und kleinen organischen Molekülen.
Bei der Gas-Flüssig-Chromatographie wird die flüssige Phase auf ein Trägermaterial aufgebracht. Dabei beruht die Trennung auf der Verteilung der gasförmigen Probensubstanz zwischen dem Flüssigkeitsfilm und der mobilen Phase, weshalb sie auch Verteilungschromatographie genannt wird. Die stationäre Phase kann zum Beispiel ein Kieselgel mit einem derivatisierten Flüssigkeitsfilm sein.42, 43
Kapillarsäulen
Die Kapillarsäulen besitzen im Gegensatz zu den gepackten Säulen keine Säulenfüllung. Das heißt sie haben einen Hohlraum, der den Säulenwiderstand verringert. Aus diesem Grund können diese Säulen eine Länge von 25 bis 100 Meter besitzen. Je größer die Schichtdicke der Trennflüssigkeit in der Kapillare ist, desto größer ist ihre Kapazität und desto geringer ist ihre Trennleistung.44, 45
Sie haben die Trennschicht nur auf der äußeren Seite, wobei man sie in drei Arten unterteilen kann:
- PLOT-Säule (engl. porous layer open tubular column)
- SCOT-Säule (engl. support-coated open tubular column)
- WCOT-Säule (engl. wall-coated open tubular column)
PLOT-Säulen werden auch Schichtkapillare genannt. Bei ihnen befindet sich das feste Trägermaterial direkt an der Kapillarwand und eignet sich für Analyten, die nicht an einer flüssigen stationären Phase separiert werden können (zum Beispiel kurzkettige Kohlenwasserstoffe).
SCOT-Säulen sind trägerbeschichtete Kapillaren, das heißt, sie besitzen ein festes Trägermaterial mit einem Flüssigkeitsfilm, der auf die Kapillarwand aufgetragen ist. Sie eignen sich für leicht flüchtige Analyten, die auf den PLOT-Säulen zu stark zurückgehalten werden.
WCOT-Säulen bestehen aus einem dünnen Flüssigkeitsfilm, der auf die Kapillarwand aufgetragen wurde. Aus diesem Grund werden sie auch Dünnfilm-Kapillare genannt. Verglichen mit anderen Kapillarsäulen besitzen sie die größte Trennleistung und finden somit Einsatz in der Spurenanalytik.46
Detektoren
Um die aus der Trennsäule eluierten Substanzen zu erkennen und zu registrieren, werden Detektoren verwendet. Diese sollten dabei eine hohe Empfindlichkeit und einen großen linearen Bereich bei der Signalintensität aufweisen, um präzise quantitative Bestimmungen zu erfassen.47
Man unterscheidet zwischen konzentrations- und massenstromabhängigen Detektoren. Das Signal von konzentrationsabhängigen Detektoren ist vom Volumen des Trägergases abhängig, beim massenstromempfindlichen Detektor ist das Signal dafür unabhängig vom Trägergasvolumen.
Die Wahl des jeweiligen Detektors hängt dabei von den Eigenschaften der nachzuweisenden Substanz ab. Mit der Wahl des Detektors wird auch das Einsetzen des Trägergases festgelegt.
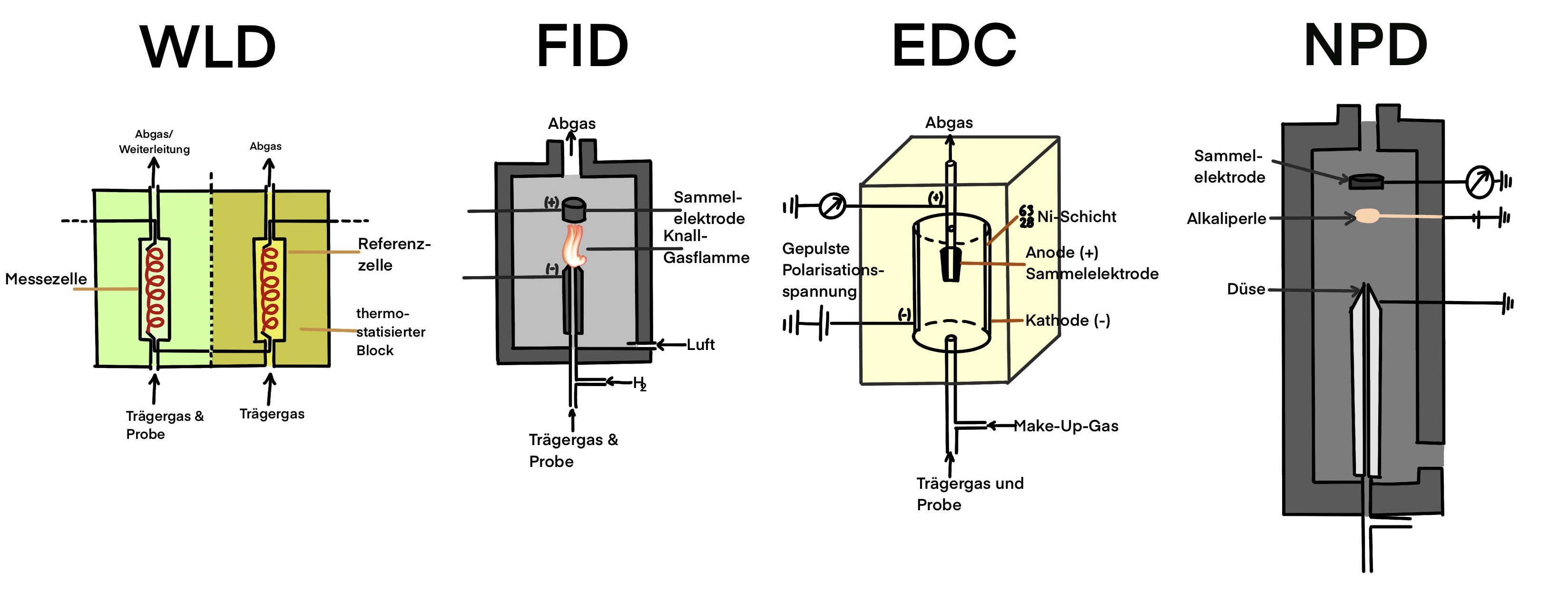
Wärmeleitfähigkeitsdetektor WLD (konzentrationsabhängig)
Bei dem WLD wird die Änderung der Wärmeleitfähigkeit erfasst, die durch die Elution der Substanzen im Vergleich zum reinen Trägergas entsteht. Hier wird der von der Säule kommende Gasstrom an einem Heizdraht (Filament) oder Halbleiterelement (Thermistor) vorbeigeführt. Diese Bestandteile werden durch das Trägergas konstant leicht abgekühlt. Da die eluierenden Analysensubstanzen eine niedrigere Wärmeleitfähigkeit als das Trägergas besitzen, werden die Messelemente folglich weniger abgekühlt. Diese Temperaturerhöhung wird als elektrische Widerstandsänderung gegenüber dem Referenzelement gemessen und als Signal registriert.
Beim WLD wird ein Trägergas mit besonders hoher Wärmeleitfähigkeit (H2, He) verwendet, um die Empfindlichkeit zu steigern. Allgemein ist der WLD nicht so empfindlich wie andere Detektoren, jedoch universell einsetzbar. Substanzen wie z.B. H2O, CO2, N2 lassen sich mithilfe des Detektors erfassen.49
Flammenionisationsdetektor FID (massenstromabhängig)
Beim FID werden die aus der Säule kommenden Substanzen in einer Knallgasflamme verbrannt. An der Düse und Spitze der Flamme befinden sich Elektroden. Die bei der Verbrennung entstandenen Radikale zerfallen zu Ionen, welche dann zwischen den Elektroden einen Stromfluss erzeugen. Dieser Stromfluss wird als Signal registriert. Es handelt sich somit um einen destruktiven Detektor, der nicht für präparative Anwendungen geeignet ist.
Der FID besitzt eine sehr hohe Empfindlichkeit und einen großen Linearitätsbereich, der wichtig für quantitative Bestimmungen ist. Während der Quantifizierung muss jedoch auf einen konstanten Fluss der mobilen Phase geachtet werden, um die Peakfläche nicht zu vergrößern. Außerdem kann der FID universell für organische Substanzen eingesetzt werden, da die zu der Signalerzeugung benötigten Ionen hauptsächlich aus der Verbrennung von organischen Kohlenwasserstoff-Gruppen entstehen. 50
Beim Trägergas gibt es so gut wie keine Einschränkungen. Jedoch sollte der Verwendung von H2 beachtet werden, dass die Empfindlichkeit des Detektors nicht durch Veränderung der Flammentemperatur gestört wird. 51
Elektroneneinfangdetektor ECD
Der ECD besteht aus einer Ionisationskammer, in der sich ein β-Strahler (3H, 63Ni), sowie eine Kathode und Anode befinden. Verbindungen mit einer hohen Elektronenaffinität (z.B. halogenhaltige Substanzen) bilden in dieser Kammer durch Elektroneneinfang negative Ionen. Durch die Elektronenaufnahme der eingsetzten Probe wird der vorhandene Ionisationsgrundstrom (Ionisierung vom Trägergas als „Grundstrom“) verringert, wodurch das Detektorsignal dargestellt wird.
Einen besonderen Einfluss auf die Ionisierung besitzt das Trägergas: Verwendet wird Stickstoff oder Argon unter Zusatz von 5 Vol.-% Methan. Dabei wird Methan nicht ionisiert, sondern dient viel mehr als Puffer, damit störende Nebenreaktionen verhindert werden.
Der ECD ist sehr selektiv und wird vor allem aufgrund seiner hohen Empfindlichkeit gegenüber halogenhaltigen Verbindungen genutzt. Die wesentlichen Nachteile dieses Detektors sind die gesonderten Richtlinien aufgrund der Radioaktivität, sowie die Anforderungen an ein hochreines Trägergas.52
Thermionischer Detektor (TCD; NPD)
Der NPD - Stickstoff-Phosphor-Detektor - (eine Art des TCDs) gehört zu den selektiven Detektoren, da er eine hohe Element- und Verbindungsspezifität für Phosphor und Stickstoff besitzt.
Sein Aufbau gleicht dem des FIDs, wobei er zusätzlich eine beheizte Alkalisalzperle (z.B aus RbCl) besitzt. An deren Oberfläche werden bevorzugt stickstoff- und phosphorhaltige Verbindungen ionisiert. Dabei ist die Flammentemperatur niedriger als bei dem FID, sodass Kohlenwasserstoffe nicht ionisiert werden und die Selektivität nicht verringert wird. 53
Der Vorteil des NPDs ist seine hohe Empfindlichkeit und Selektivität. Die Nachteile sind die Destruktion der Probe, der Verschleiß der Alkaliperle aufgrund halogenidhaltiger Substanzen und die Abtrennung von Probenbestandteilen, die bei der Verbrennung feste Verbrennungsrückstände bilden. Seine Rolle wird häufiger durch die MS-Kopplung (s. 2.4.5) ersetzt. 54
Massenselektiver Detektor (GC-MS)
Massenselektive Detektoren werden mit einer Gaschromatographie/Massenspektrometrie-Kombination vereinigt. Mittels der Kombination der Gaschromatographie mit der Massenspektrometrie besteht die Möglichkeit, jede gaschromatographierbare Verbindung zu detektieren, zu quantifizieren und gleichzeitig für jede Substanz charakteristische Informationen zu erhalten, die zur raschen und sicheren Identifizierung der Verbindung führen können. 55
Bei diesem Detektor wird das Säuleneluat in die Ionenquelle des Massenspektrometers (MS) geleitet. Die jeweiligen Ionisationsarten werden im Massenspektrometrie-Artikel Massenspektrometrie näher erklärt. Durch die Ionisierung werden die Moleküle der Substanzen entweder zertrümmert oder protoniert und liegen dann meist als einfach geladene Ionen vor. Im folgenden Analysator werden die Ionen durch angelegte elektrische und/oder magnetische Felder nach ihrem Masse-zu-Ladung-Verhältnis getrennt und in regelmäßigen Zeitabständen Massenspektren aufgenommen und elektronisch gespeichert.
Nach dem Analysator folgt der Detektor. In diesem Fall können Photomultiplier, Sekundärelektronenvervielfacher (SEV), Faraday-Auffänger, Daly-Detektoren, Mikrokanalplatten (MCP) oder Channeltrons eingesetzt werden.56
Einzelnachweise
1 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/croma/gc_detail1.vlu.html 22.05.2021 14:00 ⇑
2 https://www.chemie.de/lexikon/Gaschromatographie.html#:~:text=Der%20Vorteil%20besteht%20in%20der,Analysen%20deutlich%20gesenkt%20werden%20kann 22.05.2021 14:30 ⇑
3 https://studyflix.de/chemie/gaschromatographie-1602^ 22.05.2021 15:00 ⇑
4 G.Rücker, M.Neugebauer, G.G.Willems, Instrumentelle pharmazeutische Analytik, WVG mbH Stuttgart, S.416 ⇑
5 G.Rücker, M.Neugebauer, G.G.Willems, Instrumentelle pharmazeutische Analytik, WVG mbH Stuttgart, S.416,f. ⇑
6 G.Rücker, M.Neugebauer, G.G.Willems, Instrumentelle pharmazeutische Analytik, WVG mbH Stuttgart, S.434 ⇑
7 Abbildung: Charlotte Kalinowski - GoodNotes - Inspiration Skript zum Seminar HPLC, Francke Nadine ⇑
8 https://www.lernhelfer.de/schuelerlexikon/chemie/artikel/gaschromatografie#:~:text=Die%20Zeit%2C%20die%20eine%20Substanz,Komponenten%20ergibt%20sich%20ein%20Gaschromatogramm. 22.05.2021 16:30 ⇑
9 Abbildung: Charlotte Kalinowski - GoodNotes - Inspiration Skript zum Seminar HPLC, Francke Nadine ⇑
10 N.Francke, Seminar zum Praktikum Instrumentelle Analytik, HPLC, S.20-24 ⇑
11 https://www.chemie.de/lexikon/Van-Deemter-Gleichung.html 22.05.2021 17:00 ⇑
12 https://de.wikipedia.org/wiki/Van-Deemter-Gleichung 22.05.2021 17:00 ⇑
13 S.Bendas, Seminar zum Praktikum Instrumentelle Analytik, Gaschromatographie S.28-33 ⇑
14 G.Rücker, M.Neugebauer, G.G.Willems, Instrumentelle pharmazeutische Analytik, WVG mbH Stuttgart, S.440 ⇑
15 https://www.chemie.de/lexikon/Interner_Standard.html 22.05.2021 17:05 ⇑
16 S.Bendas, Seminar zum Praktikum Instrumentelle Analytik, Gaschromatographie, S.28-33 ⇑
17 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/altlasten/analytik.vlu/Page/vsc/de/ch/3/anc/altlasten/altlast_2/altlast_2_5/altlast_2_5_1/gc_m870902.vscml.html 22.05.2021 18:00 ⇑
18 https://www.chemie.de/lexikon/Gaschromatographie.html#:~:text=Der%20Vorteil%20besteht%20in%20der,Analysen%20deutlich%20gesenkt%20werden%20kann 22.05.2021 18:00 ⇑
19 G.Rücker, M.Neugebauer, G.G.Willems, Instrumentelle pharmazeutische Analytik, WVG mbH Stuttgart, S.417 ⇑
20 G.Rücker, M.Neugebauer, G.G.Willems, Instrumentelle pharmazeutische Analytik, WVG mbH Stuttgart, S.417 ⇑
21 G.Rücker, M.Neugebauer, G.G.Willems, Instrumentelle pharmazeutische Analytik, WVG mbH Stuttgart, S.440 ⇑
22 G.Rücker, M.Neugebauer, G.G.Willems, Instrumentelle pharmazeutische Analytik, WVG mbH Stuttgart, S.440 ⇑
23 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/altlasten/analytik.vlu/Page/vsc/de/ch/3/anc/altlasten/altlast_2/altlast_2_5/altlast_2_5_1/gc_m870902.vscml.html^ 22.05.2021 17:30 ⇑
24 Abbildung: Tatiana Gorpishina - Procreate - Inspiration https://de.wikipedia.org/wiki/Gaschromatographie 21.05.2021 12:03 Uhr ⇑
25 „Instrumentelle Analytik“ Andreas Dominik, Dieter Steinhilber, Deutscher Apotheker Verlag Stuttgart 2002, 2.Auflage S.177 ⇑
26 „Instrumentelle Analytik“ Andreas Dominik, Dieter Steinhilber, Deutscher Apotheker Verlag Stuttgart 2002, 2.Auflage S.177 ⇑
27 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/croma/gaschromatographie_1.vlu/Page/vsc/de/ch/3/anc/croma/gc/mob_phase/mobphasgcm64ht0600.vscml.html 20.05.2021 15:52 Uhr ⇑
28 „Instrumentelle Analytik“ Andreas Dominik, Dieter Steinhilber, Deutscher Apotheker Verlag Stuttgart 2002, 2.Auflage S.177 ⇑
29 Abbildung: Tatiana Gorpishina - Procreate - Inspiration https://www.geochemie.ifg.uni-kiel.de/de/lehrmaterialien/bachelor/sommersemester/mnf-geow-b201-v-geochemische-analytik/kapitel-7-analytische-chromatographie-gc-ms-2020.pdf 21.05.2021 12:05 Uhr ⇑
30 „Instrumentelle Analytik“ Andreas Dominik, Dieter Steinhilber, Deutscher Apotheker Verlag Stuttgart 2002, 2.Auflage S.178 ⇑
31 https://www.chemie-schule.de/KnowHow/Split/Splitless-Injektor 22.05.2021 10:03 Uhr ⇑
32 hhttps://phenomenex.blob.core.windows.net/documents/50b8ec34-2110-4c38-844d-164744d012f3.pdf 16.06.2021 10:30 Uhr ⇑
33 https://www.hlnug.de/fileadmin/dokumente/altlasten/handbuch/hba35_web.pdf 16.06.2021 10:44 Uhr ⇑
34 „Instrumentelle Analytik“ Andreas Dominik, Dieter Steinhilber, Deutscher Apotheker Verlag Stuttgart 2002, 2.Auflage S.178 ⇑
35 http://www.chemgapedia.de/vsengine/tra/vsc/de/ch/3/anc/chromatographie1.tra/Vlu/vsc/de/ch/3/anc/croma/gc_probenaufgabe.vlu/Page/vsc/de/ch/3/anc/croma/gc/direkt/dschleif/dosierschleifgcm66ht0600.vscml.html 20.5.2021 16:30 Uhr ⇑
36 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/croma/gc_probenaufgabe.vlu/Page/vsc/de/ch/3/anc/croma/gc/direkt/kalt/kaltaufggc_m66ht0600.vscml.html 22.05.2021 16:45 Uhr ⇑
37 Online Vorlesung SS2021 TU-Braunschweig Fach: Vorlesung Instrumentelle Analytik Autor: hwaetzig Video: Gaschromatographie Nov 2020 Aufzeichnungsdatum: 26.11.2020 12:59 Uhr; 22.05.2021 17:30 Uhr ⇑
38 „Instrumentelle Analytik“ Andreas Dominik, Dieter Steinhilber, Deutscher Apotheker Verlag Stuttgart 2002, 2.Auflage S.178,179 ⇑
39 „https://www.chemie.de/whitepaper/126475/saeulen-fuer-die-gaschromatographie-auswahl-und-anwendung-von-gc-saeulen.html 17.06.2021 15:34 Uhr ⇑
40 „Instrumentelle Analytik“ Andreas Dominik, Dieter Steinhilber, Deutscher Apotheker Verlag Stuttgart 2002, 2.Auflage S.178,179 ⇑
41 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/croma/gc_detail1.vlu/Page/vsc/de/ch/3/anc/croma/gc/stat_phase/gepackte_saeule/gepackt1m71te0201.vscml.html 17.06.2021 15:50 Uhr ⇑
42 Online Vorlesung SS2021 TU-Braunschweig Fach: Vorlesung Instrumentelle Analytik Autor: hwaetzig Video: Gaschromatographie Nov 2020 Aufzeichnungsdatum: 26.11.2020 12:59 Uhr; 22.05.2021 17:30 Uhr ⇑
43 „Instrumentelle Analytik“ Andreas Dominik, Dieter Steinhilber, Deutscher Apotheker Verlag Stuttgart 2002, 2.Auflage S.178,179 ⇑
44 Online Vorlesung SS2021 TU-Braunschweig Fach: Vorlesung Instrumentelle Analytik Autor: hwaetzig Video: Gaschromatographie Nov 2020 Aufzeichnungsdatum: 26.11.2020 12:59 Uhr; 22.05.2021 17:30 Uhr ⇑
45 „Instrumentelle Analytik“ Andreas Dominik, Dieter Steinhilber, Deutscher Apotheker Verlag Stuttgart 2002, 2.Auflage S.178,179 ⇑
46 http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/croma/gc_detail1.vlu/Page/vsc/de/ch/3/anc/croma/gc/stat_phase/kapillarsaeule/kapillarsaeule2.vscml.html 17.06.2021 17:11 Uhr ⇑
47 E. Cremer, Gas‐Chromatographie als Methode zur Untersuchung der Eigenschaften von Grenzflächen, Adsorptionsschichten und Flüssigkeiten, Berichte der Bunsengesellschaft für physikalische Chemie, 10.1002/bbpc.19650690909, 69, 9‐10, (802-811), (2014) ⇑
48 Abbildung: Tatiana Gorpishina - Procreate - Inspiration http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/croma/gaschromatographie_1.vlu/Page/vsc/de/ch/3/anc/croma/gc/detektoren/gcdetm75ht0401.vscml.html 21.05.2021 12:10 Uhr ⇑
49 Dominik, Andreas, Dieter Steinhilber, and Mario Wurglics. Instrumentelle Analytik kompakt. Wissenschaftliche Verlagsgesellschaft mbH: Stuttgart, 2013. ⇑
50 Dominik, Andreas, Dieter Steinhilber, and Mario Wurglics. Instrumentelle Analytik kompakt. Wissenschaftliche Verlagsgesellschaft mbH: Stuttgart, 2013. ⇑
51 Dominik, Andreas, and Dieter Steinhilber. Instrumentelle Analytik: Kurzlehrbuch und kommentierte Originalfragen für Pharmazeuten. Deutscher Apotheker Verlag, 2002. ⇑
52 Dominik, Andreas, Dieter Steinhilber, and Mario Wurglics. Instrumentelle Analytik kompakt. Wissenschaftliche Verlagsgesellschaft mbH: Stuttgart, 2013. ⇑
53 Helm, Mark, and W. Stefan. Instrumentelle Bioanalytik: Einführung für Biologen, Biochemiker, Biotechnologen und Pharmazeuten. John Wiley & Sons, 2013 ⇑
54 Dominik, Andreas, Dieter Steinhilber, and Mario Wurglics. Instrumentelle Analytik kompakt. Wissenschaftliche Verlagsgesellschaft mbH: Stuttgart, 2013. ⇑
55 Helm, Mark, and W. Stefan. Instrumentelle Bioanalytik: Einfuhrung fur Biologen, Biochemiker, Biotechnologen und Pharmazeuten. John Wiley & Sons,2013. ⇑
56 Dominik, Andreas, and Dieter Steinhilber. Instrumentelle Analytik: Kurzlehrbuch und kommentierte Originalfragen für Pharmazeuten. Deutscher Apotheker Verlag, 2002. ⇑
Monographiebeispiel: Desfluran (Prüfung auf Reinheit)
Stoffcharakterisierung (Wirkung und Anwendung)
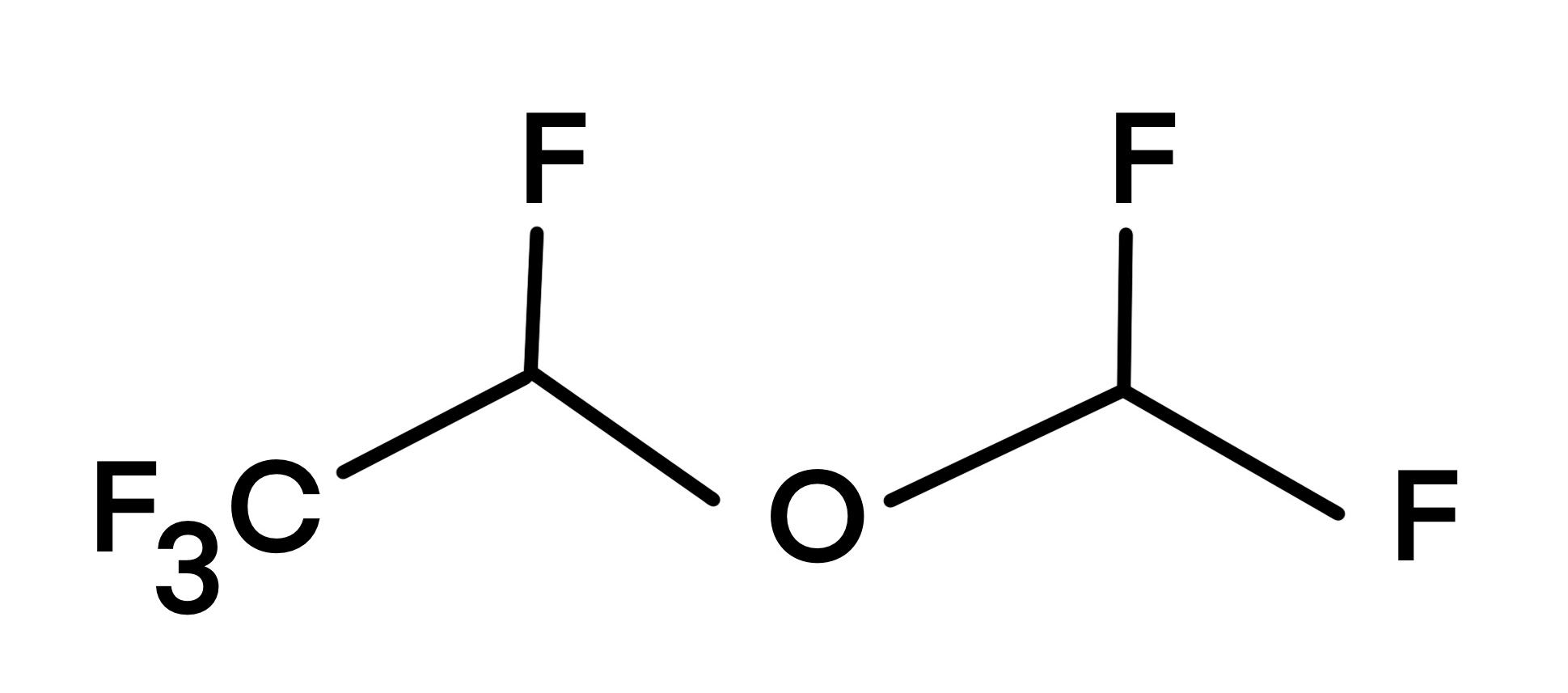
Desfluran (IUPAC: (2RS)-2-(Difluormethoxy)-1,1,1,2-tetrafluorethan) ist eine klare, farblose und leicht riechende Flüssigkeit. Sie ist kaum löslich in Wasser und mischbar mit wasserfreiem Ethanol. Die Siedetemperatur liegt bei etwa 22 °C. 2
Desfluran gehört zur Gruppe der halogenierten Methylether (s. Bild) und zur Wirkstoffklasse der Flurane (verwandt mit Sevofluran und Isofluran). 3 Bei diesem Medikament handelt es sich um ein Inhalationsanästhetikum, welches genutzt wird, um eine Allgemeinnarkose einzuleiten oder diese aufrecht zu erhalten. Die Wirkung ist stark hypnotisch, denn nach der Einnahme werden das Bewusstsein und das Schmerzempfinden reversibel gehemmt. Außerdem wird die willkürliche Motorik unterdrückt und die Atmung sowie das Herz-Kreislauf-System werden gedämpft. 4
Die Wirkung des Medikaments ist durch schnelles An- und Abfluten des Stoffes gut zu kontrollieren, da es nur wenig löslich in Wasser ist. Es besitzt mit etwa 0,4 einen niedrigen Blut/Gas-Verteilungskoeffizienten. 5, 6 Dieser beschreibt das Verhältnis der Konzentration von Desfluran im Blut zur Konzentration in der Alveolarluft.7 Durch den niedrigen Blut/Gas-Verteilungskoeffizienten lassen sich schnelle Einschlaf- und Aufwachphasen erzielen, weshalb es auch in der ambulanten Chirurgie eingesetzt wird. Die Ausscheidung von Desfluran findet größtenteils unverändert über die Lungen statt und es wird nur in geringem Maße verstoffwechselt. 8
Durchführung der Monographie
Das Arzneibuch sieht für die Prüfung auf Reinheit von Desfluran bei verwandten Substanzen eine Prüfung mittels Gaschromatographie vor.
Bei der gaschromatographischen Prüfung auf Reinheit dient die Substanz als Untersuchungslösung. Sie muss auf eine Temperatur unterhalb von
10 °C abgekühlt sein. Die Prüfungen erfolgen bei einer Temperatur unterhalb von 20 °C.
Folgende Referenzlösungen werden für die Durchführung der Monographie verwendet:
- Referenzlösung a: Es wird Substanz mit Desfluran-Verunreinigung A CRS und Isofluran CRS (Verunreinigung B) in einer 50-ml-Flasche, die mit einem Septum verschlossen ist, vermengt. Zu dieser Mischung wird Aceton R (Verunreinigung H), Chloroform R (Verunreinigung F) und Dichlormethan R (Verunreinigung E) mit einer gasdichten Spritze versetzt und die Substanz wird mit der Lösung verdünnt.
- Referenzlösung b und c: Bei den Referenzlösungen b und c wird jeweils die vorherige Referenzlösung verdünnt.
Alle drei entstandenen Lösungen werden bei einer Temperatur unter 10 °C aufbewahrt.
Es werden immer 2 µl von den Referenzlösungen eingespritzt. 9
Als geeignetes Trägergas wird bei der Reinheitsbestimmung Helium zur Chromatographie R verwendet. Die Durchflussrate beträgt 2,0 ml • min -1 und das Splitverhältnis 1:25. Die Chromatographiedauer beträgt 35 Minuten und zur Detektion wird die Flammenionisation verwendet. 10
Säule
Die Säule bei der Prüfung auf Reinheit von Desfluran besteht aus Quarzglas. Sie hat eine Länge von 105 m und einen Durchmesser von 32 mm. Zudem besteht die stationäre Phase aus Trifluorpropylmethylpolysiloxan R und weist eine Filmdicke von 1,5 µm auf. 11 Eine geeignete Säule für die gaschromatographische Prüfung bei Desfluran ist die Rtx-200. 12 Sie besitzt eine gute thermische und chemische Stabilität und ist in der Lage viele schwierige Trennungen auszuführen. 13
Temperaturen
| Gerät | Temperatur |
|---|---|
| Säule | 30°C |
| Probeneinlass | 150°C |
| Detektor | 200°C |
Verunreinigungen
Die Probe wird gaschromatographisch auf verschiedene Verunreinigungen wie z.B. Trichlormethan, Chloroform und Aceton geprüft. 14
Des Weiteren sieht das Arzneibuch bei sauer oder alkalisch reagierenden Substanzen eine Prüfung mit Bromcresolpurpur (Indikator) vor.
Zudem kann eine Prüfung auf Fluorid mithilfe der Potentiometrie erfolgen, wobei höchstens 10 ppm vorhanden sein dürfen. Die Fluorid-Ionen können bei der Herstellung entstanden sein. Die Prüfung muss erfolgen, da Desfluran mit Fluorid-Ionen reagieren kann und später ein Nachreinigungsprozess erfolgen müsste. 15 Außerdem wird auf nicht flüchtige Substanzen mit Hilfe von Stickstoff geprüft. 16
Auswertung/ Interpretation und Eignung der analytischen Methode
Auswertung
Nach einer Chromatographiedauer von ca. 35 Minuten können die relativen Retentionszeiten der Verunreinigungen (bezogen auf Desfluran; tR: 11,5 min) über das Chromatogramm ausgewertet werden. Mithilfe der relativen Retentionszeiten kann eine Aussage darüber getroffen werden, ob Verunreinigungen enthalten sind und um welche es sich handelt. Für die einzelnen Substanzen gibt es verschiedene Grenzwerte, die laut Arzneibuch eingehalten werden müssen. Die quantitative Bestimmung erfolgt über die Peakflächen der einzelnen Komponenten, wobei die Peaks der Verunreinigungen mit den Peaks der Referenzlösungen verglichen werden. 17
Eignung und Interpretation
Zur Bestimmung der Reinheit wurde bei dieser Monographie eine chromatographische Methode gewählt, welche es erlaubt auch die einzelnen Verunreinigungen quantitativ mittels der Peakflächen auszuwerten. Die Gaschromatographie eignet sich hier besonders gut, da Desfluran eine Siedetemperatur von ca. 22 °C hat und somit leicht in die gasförmige Phase überführbar ist. Bei den möglichen Verunreinigungen handelt es sich um Aceton, halogenierte Ether sowie Halogenkohlenwasserstoffe und insofern um verwandte Substanzen des Desflurans. Unter den gegebenen Bedingungen sind auch diese mittels Gaschromatographie bestimmbar. Zudem eignet sich der Flammenionisationsdetektor bei dieser Reinheitsbestimmung, da die bedeutsamen Substanzen oxidierbar sind und sie somit in der Knallgasflamme des Detektors verbrannt werden können.
Einzelnachweise
1 Abbildung: Tatiana Gorpishina - Procreate ⇑
2 Ph. Eur. 10.0/1666 Desfluran ⇑
3 https://medlexi.de/Desfluran, 16.06.2021 15:06 ⇑
4 https://www.fachinfo.de/suche/fi/007952, 16.06.2021 15:07 ⇑
5 Arzneibuch Kommentar Desfluran 9.8/1666 ⇑
6 https://medlexi.de/Desfluran, 16.06.2021 15:06 ⇑
7 https://de.wikipedia.org/wiki/Inhalationsan%C3%A4sthetikum#:~:text=Das%20Verh%C3%A4ltnis%20der%20Konzentration%20des,Kompartimenten%20die%20gleiche%20Konzentration%20herrscht, 17.06.2021 21:08 ⇑
8 https://www.pharmawiki.ch/wiki/index.php?wiki=Desfluran, 16.06.2021 15:10 ⇑
9 Ph. Eur. 10.0/1666 Desfluran ⇑
10 Ph. Eur. 10.0/1666 Desfluran ⇑
11 Ph. Eur. 10.0/1666 Desfluran ⇑
12 Arzneibuch Kommentar Desfluran 9.8/1666 ⇑
13 https://www.restekgmbh.de/produkte/saeulen/rtxr-200, 17.06.2021 18:39 ⇑
14 Ph. Eur. 10.0/1666 Desfluran ⇑
15 Arzneibuch Kommentar Desfluran 9.8/1666 ⇑
16 Ph. Eur. 10.0/1666 Desfluran ⇑
17 Ph. Eur. 10.0/1666 Desfluran ⇑
TU-Braunschweig Institut für Medizinische und Pharmazeutische Chemie Seminar: Instrumentelle Analytik Kontakt: tubs@t-kellner.de
